Liquid chromatography
Published over 5 years ago. See the latest and most current information on Liquid chromatography.
The implementation of ultrafast liquid chromatography techniques to reduce method time and increase analytical throughput has grown tremendously in recent years. Instruments capable of higher pressures and flow rates, coupled with modern column technologies that can provide sufficient chromatographic efficiencies even at very short lengths, have provided the means to drastically increase separation speeds. Improvements to detector modules and new data processing algorithms are enabling effective signal acquisition at the requisite rates. The rate of autosampler operation has also increased, but has lagged behind these other advancements and now represents one of the primary bottlenecks for further increases to sample throughput in LC [1]. In this overview, current injector and autosampler technology will be discussed, along with recent strategies that have been used to reduce injection cycle time and how these might be further developed in coming years.
In a typical method sequence that is set up within a chromatography data system (CDS), a series of runs are listed that detail which vial is to be sampled and which chromatographic method is to be used for each injection. In a standard instrument operating mode, the autosampler sequence occurs before each injection. This sequence typically includes movement of the needle to the designated sample vial position, a sample draw, movement back to the injector valve (if a needle seat port is used), and a series of needle washes to reduce sample carryover [2, 3]. This approach has multiple limitations that can increase instrument cycle time and thus reduce overall analytical throughput. The primary issue is the fact that the autosampler sequence and the chromatographic run are performed asynchronously, which means that the total instrument cycle time is the sum of both operation times rather than simply the slower of the two. The simple fix to this limitation is to simultaneously perform the autosampler sequence with the preceding chromatographic run so that as soon as one chromatographic analysis is finished, the next can begin immediately because the sample has already been delivered to the injector. Such functionality is often available as a setting of modern CDSs, using functions such as ‘PrepareNextInjection’ [4], ‘Enable Overlapped Injection’ [5], or “Prep Ahead” [6] within the autosampler control code. This approach was demonstrated for the analysis of over-the-counter (OTC) analgesic compounds with a complete chromatographic cycle time of 20 s (total 5 min analysis time for 15 replicate runs) [7]. The short separation time was achieved by operating a 2.1 x 50 mm column packed with sub-3 µm core-shell particles at 1.3 mL/min, which required UHPLC instrumentation as the column pressure exceeded 750 bar. The autosampler sequence time was reduced to less than 13 s by limiting the time of the needle wash processes, which ensured that it could be completed prior to the finish of the preceding 20 s chromatographic run (Figure 1). Although no significant issues were observed with carryover in this example, it can become an issue depending on the specific analytes and sample solvent coming into contact with the autosampler needle and loop. In a demonstration of a more robust, qualified high-throughput OTC analgesic assay, a 40 s cycle time was achieved combining an equivalent separation time and a ~30 s autosampler sequence that included needle washes [8].
Isocratic separations were used in both of the examples described in the previous paragraph. With synchronous operation of the autosampler and chromatographic analysis, the primary concern is to ensure that no valve actuation steps are coincident with eluting peaks. Such actuations can result in pressure changes that lead to detector signal disruptions and distortions to observed peak shape. When rapid (or ‘ballistic’) gradient separations are used in low cycle time analyses, the system dwell volume is another critical consideration because it affects how long it takes the programmed gradient to reach the column [9]. Connections between the pump and injector (which may include an in-line mixer), flow paths within the injector itself, and additional connections to the column all contribute to this value. Autosampler configurations that do not flow through the needle typically provide smaller volumes that are more amenable for high-throughput gradient methods, though their design creates the potential for air bubble formation in the injector flow path [3].
A variation on synchronous injection cycle sample preparation is the multiple injections in a single experimental run (MISER) technique [10]. In MISER, multiple injections are run in the same way that is described above, but the data is not collected as individual chromatograms and is instead collected in a single data file. This is an ideal strategy for qualitative work in which a rapid comparison of a series of runs in an injection sequence can quickly identify general trends and outliers (i.e. high and low abundance peaks) within a sample set. For example, MISER has been used to rapidly compare the amount of hop-derived components in different varieties of beer [11], caffeine concentration in beverages [12], and capsaicin content in peppers and hot sauces [13]. It is also a powerful strategy for the enantiopurity monitoring and other high-throughput screening needs within the pharmaceutical industry [1, 14]. The key drawback in MISER is the increased difficulty in peak quantification compared to collecting individual chromatograms for each unique run. Many software algorithms for quantitation use area comparisons between analyte and internal standard peaks at a specific retention time (and/or m/z value, depending on the detection mode), which can be more difficult to incorporate when a number of individual separations are all plotted on a single chromatogram. While it can still be accomplished through chromatogram splitting programs [9] or manual processing, these strategies require more user interaction to ensure all the peaks are properly matched; the overall workload increases significantly as the number of runs grows in a high-throughput screening experiment. Therefore, it is important to consider the overall purpose of the high-throughput experiments being conducted, more specifically whether they are qualitative or quantitative, in order to determine which approach is most appropriate in efforts to reduce cycle time.
A modified version of the synchronous autosampler cycle approach is to actually inject the subsequent sample during a preceding run rather than to just prepare for injection. This approach is often used when performing ‘stacked injections’ for chiral purifications in preparative-scale HPLC or SFC. When isocratic elution conditions are used and the peak elution window is short relative to the total peak elution time, stacked injections reduce solvent consumption and improve production rates [15]. The approach is also effective with non-retentive chromatography modes (i.e. size exclusion chromatography, hydrodynamic chromatography) [16, 17], where the finite elution window of the peaks is known based on the volume difference between analytes at the high and low ends of the size range for a given stationary phase. Because of the initial delay between sample injection and the first eluted peak, an additional injection can be performed during this time gap so that this same first peak from the second injection elutes closely to the final peak from the initial sample. This was demonstrated for a preparative-scale hydrodynamic chromatography separation used to reduce the particle size distribution of a silica packing material [18,19]. Because multiple injections were needed to obtain the amount of purified sample that was required, this strategy eliminated any added time between runs used to deliver the sample to the injection loop [20] (Figure 2). A similar approach has also been described for aggregate characterisation of monoclonal antibodies [21, 22]. Although these examples included slightly longer runs and the added time would have been only a fraction of the overall cycle time, the technique would have an impact on high-throughput size-based separations [23]. A similar process has been developed for retentive chromatography modes with more complex mixtures, but it can be much more difficult to implement due to the optimisation required to avoid peak co-elutions [24]. In those instances, the previously described strategies may be preferred.
The use of columns with smaller dimensions, and thus volumes, has made the consideration of instrument-based broadening effects a critical aspect of high-throughput LC. The most basic description of broadening due to the injection cycle is based solely on the volume of sample injected [25]:
(1)
However, this is a simplification that assumes a rectangular injection band profile and is not a true estimation of the actual broadening that occurs due to the injection process [26]. A number of studies have identified additional causes of broadening in the injection process, many of which are flow-dependent and can grow as the separation speed is increased. In one experiment, a miniaturised electrochemical detector was used to track injector band profiling in a nano-volume internal loop design coupled a variety of tubing diameters and lengths. It was determined that the eluted peaks contained both a Gaussian and exponential component, the exponential component of which grew with increasing flow rate (Figure 3) [27]. The likely source of this broadening was the abrupt changes in channel diameter that existed between the stator, rotor, and connected tubing, where each channel mismatch created a ‘mixing chamber’. This experimental observation was further supported through fluid dynamic modelling of a computational reconstruction of the injector flow path. To reduce this broadening contribution, the tailed portion of the peak can be removed from the injection profile by performing a partial loop ‘timed pinch injection’ with rapid actuation of the valve back to the ‘load’ position after injection (Figure 3), although this can reduce injection repeatability compared to traditional full loop injection [27].
Commercial autosamplers in analytical-scale systems have also been studied to determine the broadening effects due to injection. In systems that contain a capillary connection between the needle seat and the injector (common for flow-through needle designs), smaller-diameter tubing is critical to minimise broadening [25, 28]. There was also an observable increase in the injector-based broadening with increasing flow rates, which levelled off around 0.8 mL/min [28]. For loop-based injectors that are more similar to those that were investigated in the preceding paragraph, the impact of both injection volume and flow rate on the observed broadening were both lower than with the flow-through needle injector. However, with larger inner diameter loops, added contributions at high flow rates were also observed in this format [28]. Fluid dynamic modelling was also used to further explore these observations, and the presence of two regimes within the injector were identified: the convective regime and the diffusion regime [29]. The dominant regime depends on a number of variables, including the injection volume, needle diameter (specifically for flow-through needle injectors), analyte diffusion coefficient, and chromatographic flow rate. Under typical LC operating conditions that would be used for chromatographic analysis, loop-based injectors produce narrower injection bands than flow-through designs [29]. These various studies all indicate that band broadening contributions due to injection are usually underestimated and that there is typically an increase in this broadening at the higher flow rates used in high-throughput LC. Thus, further optimisation of channel structure and overall injector design are still needed to ensure high chromatographic performance in rapid LC separations.
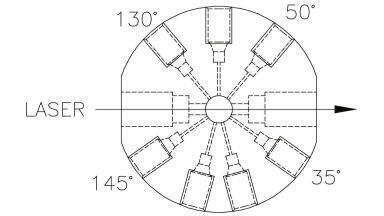
As described above, the modern injection process has been improved significantly, with the fastest autosampler cycle times now down to 7 s [17]. However, as ultrafast subsecond separations become more feasible [30], 7 s will still be far too slow to achieve high-throughput screening rates that compare to MS-based techniques which can now exceed 30 samples per second [31]. One approach is to use parallel sampling that introduces multiple samples into the flow stream simultaneously [32]. To some degree, this has already been incorporated into modern autosamplers that contain dual needle designs [33, 34], although this multiplex approach would need to be increased significantly to accommodate more rapid separations. The potential to use segmented flow droplets in a multiplex array to further increase sample throughput was described in a presentation at Pittcon 2020 [35]. The highest analysis rates that have been achieved when coupling droplets to a separation technique thus far rely upon microchip electrophoresis [36, 37, 38], as there are many successful approaches to injecting droplet streams into an electrophoretic separation channel [39]. This process can be more challenging with pressure-driven LC separations. A swan probe approach has been used to collect sample from spatial droplet arrays and deliver it to an in-line monolithic column [40], although it relies upon a channel sealing technique that limits operating pressure to 25 bar and is no faster than state-of-the-art commercial LC autosamplers. In a reaction optimisation platform that included on-line LC-MS analysis, droplet streams segmented with an inert gas were coupled directly to an injection loop with an additional vacuum port used to clear remaining liquid in the loop and reduce carryover [41]. This latter technique may have the best capacity for increased throughput, but it is critically important to precisely time the valve actuation so that only sample is injected and not the carrier phase. This may only cause minor issues if the droplets are segmented with inert gas, but could be much more detrimental when a fluorinated oil that could interfere with the separation and/or cause carryover problems is used.
As the need for fast screening methods that rely upon chromatographic separations continues to increase, the speed of every aspect of the analytical technique must be examined. Novel strategies that employ higher throughput sample introduction will be a critical aspect of the next generation of rapid measurement tools. Whether the next generation of sample injection is based upon parallel sampling, segmented flow, or some other technique is yet to be identified, most practitioners of ultrafast chromatography agree that it is a major bottleneck to be overcome. For now, the advances that have brought autosampler cycle times below 10 seconds still represent an outstanding improvement from previous instrument generations and can still facilitate higher throughput analysis in nearly any lab that relies upon LC.
1. Zawatzky, K., Barhate, C. L., Regalado, E. L., Mann, B. F., Marshall, N., Moore, J. C., Welch, C. J., Overcoming “speed limits” in high throughput chromatographic analysis. J. Chromatogr. A 2017, 1499, 211–216.
2. De Vos, J., Broeckhoven, K., Eeltink, S., Advances in Ultrahigh-Pressure Liquid Chromatography Technology and System Design. Anal. Chem. 2016, 88, 262–278.
3. Paul, C., Steiner, F., Dong, M. W., HPLC Autosamplers : Perspectives, Principles, and Practices. LC-GC North Am. 2019, 37, 514–529.
4. Neubauer, M., A Multi-Tasking Software Approach to Increasing Productivity and Throughput in UHPLC. Technical Note 172, 2016.
5. Fandiño, A., Schuette, S., Ultra-Fast Liquid Chromatography Using the Agilent 1100 Series HPLC System and 1.8-Μm ZORBAX SB C18 Rapid Resolution HT Columns. Technical Note 5989-1603EN, 2004.
6. Taylor, A. M., Jarvis, M. J. Y., Cabrices, O. G., A High Throughput Automated Sample Preparation and Analysis Workflow for Comprehensive Forensic Toxicology Screening Using LC/MS/MS. Technical Note 8130213-01, 2013.
7. Kresge, G. A., Wong, J.-M. T., De Pra, M., Steiner, F., Grinias, J. P., Using Superficially Porous Particles and Ultrahigh Pressure Liquid Chromatography in Pharmacopeial Monograph Modernization of Common Analgesics. Chromatographia 2019, 82, 465–475.
8. Kresge, G. A., Grosse, S., Zimmer, A., Grinias, K. M., De Pra, M., Wong, J. T., Steiner, F., Grinias, J. P., Strategies in developing high‐throughput liquid chromatography protocols for method qualification of pharmacopeial monographs. J. Sep. Sci. 2020, 43,
2964-2970.
9. Gilar, M., McDonald, T. S., Gritti, F., Impact of instrument and column parameters on high-throughput liquid chromatography performance. J. Chromatogr. A 2017, 1523, 215–223.
10. Welch, C. J., Gong, X., Schafer, W., Pratt, E. C., Brkovic, T., Pirzada, Z., Cuff, J. F., Kosjek, B., MISER chromatography (multiple injections in a single experimental run): The chromatogram is the graph. Tetrahedron Asymmetry 2010, 21, 1674–1681.
11. Hamper, B. C., Zawatzky, K., Zhang, V., Welch, C. J., Rapid determination of humulones and isohumulones in beers using MISER LC-MS analysis. J. Am. Soc. Brew. Chem. 2017, 75, 333–338.
12. Welch, C. J., Regalado, E. L., Kress, M. H., Kraml, C., Welch, E. C., Welch, M. J., Semmelhack, H., Almstead, D., Kress, A. S., Hidalgo, N. A., MISER LC-MS analysis of teas, soft drinks, and energy drinks. LC-GC North Am. 2015, 33, 262–269.
13. Welch, C. J., Regalado, E. L., Welch, E. C., Eckert, I. M. K., Kraml, C., Evaluation of capsaicin in chili peppers and hot sauces by MISER HPLC-ESIMS. Anal. Methods 2014, 6, 857–862.
14. Welch, C. J., High throughput analysis enables high throughput experimentation in pharmaceutical process research. React. Chem. Eng. 2019, 4, 1895–1911.
15. Miller, L. M., Pinkston, J. D., Taylor, L. T., Preparative Achiral and Chiral SFC – Method Development, Stationary Phases, and Mobile Phases. In Modern Supercritical Fluid Chromatography: Carbon Dioxide Containing Mobile Phases, John Wiley & Sons, Inc. 2019.
16. Watabe, Y., Nakajima, K., Terada, H., Increased Throughput with Nexera GPC System: Overlapped Injection and Simultaneous Determination of Polymer Additives. Application News Report L537, 2019.
17. Terada, H., Koterasawa, K., Kihara, T., Increased Analysis Throughput by Overlapped Injection Using the SIL-40 Series Autosampler. Technical Note C190-E235, 2019.
18. Thompson, J. W., Lieberman, R. A., Jorgenson, J. W., Hydrodynamic chromatography for the size classification of micron and sub-micron sized packing materials. J. Chromatogr. A 2009, 1216, 7732–7738.
19. Grinias, J. P., Godinho, J. M., Lunn, D. B., Jorgenson, J. W., Evaluation of preparative hydrodynamic chromatography of silica stationary phase supports. J. Chromatogr. A 2014, 1370, 270–273.
20. Grinias, J. P., Characterization and Control of Band Broadening in Ultra-High Pressure Liquid Chromatography Columns, University of North Carolina at Chapel Hill, 2014.
21. Farnan, D., Moreno, G. T., Stults, J., Becker, A., Tremintin, G., van Gils, M., Interlaced size exclusion liquid chromatography of monoclonal antibodies. J. Chromatogr. A 2009, 1216, 8904–8909.
22. Kahle, J., Wätzig, H., Interlaced Size Exclusion Chromatography for faster protein analysis. Eur. J. Pharm. Biopharm. 2018, 126, 101–103.
23. Uliyanchenko, E., Size-exclusion chromatography - From high-performance to ultra-performance. Anal. Bioanal. Chem. 2014, 406, 6087–6094.
24. Matsuda, R., Hayashi, Y., Ishibashi, M., Takeda, Y., An information theory of chromatography. II. Application of FUMI to the optimization of overlapped chromatograms. J. Chromatogr. A 1989, 462, 23–30.
25. Gritti, F., Guiochon, G., On the minimization of the band-broadening contributions of a modern, very high pressure liquid chromatograph. J. Chromatogr. A 2011, 1218, 4632–4648.
26. Desmet, G., Broeckhoven, K., Extra-column band broadening effects in contemporary liquid chromatography: Causes and solutions. TrAC Trends Anal. Chem. 2019, 119, 115619.
27. Grinias, J. P., Bunner, B., Gilar, M., Jorgenson, J. W., Measurement and Modeling of Extra-Column Effects Due to Injection and Connections in Capillary Liquid Chromatography. Chromatography 2015, 2, 669–690.
28. Broeckhoven, K., Vanderlinden, K., Guillarme, D., Desmet, G., On-tubing fluorescence measurements of the band broadening of contemporary injectors in ultra-high performance liquid chromatography. J. Chromatogr. A 2018, 1535, 44–54.
29. Deridder, S., Desmet, G., Broeckhoven, K., Numerical investigation of band spreading generated by flow-through needle and fixed loop sample injectors. J. Chromatogr. A 2018, 1552, 29–42.
30. Patel, D. C., Wahab, M. F., O’Haver, T. C., Armstrong, D. W., Separations at the Speed of Sensors. Anal. Chem. 2018, 90, 3349–3356.
31. Kempa, E. E., Smith, C. A., Li, X., Bellina, B., Richardson, K., Pringle, S., Galman, J. L., Turner, N. J., Barran, P. E., Coupling Droplet Microfluidics with Mass Spectrometry for Ultrahigh-Throughput Analysis of Complex Mixtures up to and above 30 Hz. Anal. Chem. 2020, 92, 12605–12612.
32. Welch, C. J., Are We Approaching a Speed Limit for the Chromatographic Separation of Enantiomers? ACS Cent. Sci. 2017, 3, 823–829.
33. Grosse, S., De Pra, M., Steiner, F., Doubling the Throughput of Long Chromatographic Methods by Using a Novel Dual LC Workflow. Application Note 72601, 2018.
34. Schneider, S., Metzlaff, M., Ultrafast Analysis of Food Preservatives Using Automated Column Regeneration and Dual-Needle Injection. Application Note 5991-6150EN, 2015.
35. Welch, C. J., Ultrahigh Performance Liquid Chromatography: Key Enabling Technology for Pharmaceutical Discovery, Development, Manufacturing. Pittcon 2020. Chicago, IL 2020.
36. Guetschow, E. D., Steyer, D. J., Kennedy, R. T., Subsecond electrophoretic separations from droplet samples for screening of enzyme modulators. Anal. Chem. 2014, 86, 10373–10379.
37. Guetschow, E. D., Kumar, S., Lombard, D. B., Kennedy, R. T., Identification of sirtuin 5 inhibitors by ultrafast microchip electrophoresis using nanoliter volume samples. Anal. Bioanal. Chem. 2016, 408, 721–731.
38. Ouimet, C. M., D’Amico, C. I., Kennedy, R. T., Droplet sample introduction to microchip gel and zone electrophoresis for rapid analysis of protein-protein complexes and enzymatic reactions. Anal. Bioanal. Chem. 2019, 411, 6155–6163.
39. Liénard–Mayor, T., Taverna, M., Descroix, S., Mai, T. D., Droplet-interfacing strategies in microscale electrophoresis for sample treatment, separation and quantification: A review. Anal. Chim. Acta. 2021, 1143, 281-297.
40. Jin, D. Q., Shi, S. W., Ma, Y., Fang, Q., LC-Swan Probe: An Integrated in Situ Sampling Interface for Liquid Chromatography Separation and Mass Spectrometry Analysis. Anal. Chem. 2020, 92, 9214–9222.
41. Reizman, B. J., Jensen, K. F., Simultaneous solvent screening and reaction optimization in microliter slugs. Chem. Commun. 2015, 51, 13290–13293.