LC-MS
Published over 8 years ago. See the latest and most current information on LC-MS.
Liquid chromatography mass spectrometry (LC-MS) based multiple attribute methods (MAM) are a relatively recent development for monitoring biologic product quality attributes (PQAs) in late development, manufacturing, and quality control of biotherapeutics. Here, a new LC-MS based MAM workflow is described that is unique in its ability to not only monitor a large number of PQAs simultaneously, but also to concurrently monitor known impurities while detecting potential new impurities in an untargeted manner. Development and implementation of the workflow is straightforward with intuitive data acquisition software. Moreover, the use of a single, integrated, data processing package simplifies PQA characterisation, monitoring, quantitation, purity testing, and reporting, and further positions the workflow as a solution well suited for downstream biopharma laboratory environments.
The ultimate goal of any manufacturing facility is to deliver the highest quality product, with the highest yield, at the lowest cost, and in the shortest time frame. Biopharmaceutical manufacturers are no different, and the pressure to meet these quality, cost, and time constraints is more urgent today than ever before as product patents expire, biosimilars enter the market, and more competitors move into the biologics space. In order to maintain market position of current drugs and be first-to-market for new drugs, biopharmaceutical companies actively search for new ways to maximise overall process efficiencies that will keep manufacturing costs down, shorten time-to-market, and best protect intellectual property.
The FDA’s quality by design (QbD) approach strives to improve overall efficiencies by building quality and consistency into the manufacturing process [1]. With QbD, biopharmaceutical manufacturers gain more flexibility and less maintenance during commercial manufacturing with fewer lot failures and better consistency in product quality. In order to implement a QbD approach, the product quality attributes (PQAs) of the biopharmaceutical must first be determined prior to the design of the manufacturing process. PQAs can be product or process derived, such as deamidation, oxidation, glycosylation, or host cell proteins. Any given PQA has the potential to affect the efficacy or safety of a biotherapeutic drug. For example, a specific modification on an antibody-based drug may interfere with its ability to bind to its target, or it may increase immunogenicity or alter its clearance rate. Thus, from these PQAs, a subset of critical quality attributes (CQAs) is established that have a measurable impact on the safety and efficacy of the drug product. These CQAs are derived from a combination of prior knowledge and experimental assessment during product development and must be controlled during production and storage. The QbD approach then specifically designs the manufacturing process to ensure these CQAs are within specified ranges and not simply derived empirically from test batches.
Currently there are a multitude of assays that are performed to characterise a biotherapeutic product and monitor its quality from process development up through product release. For example, cation exchange chromatography (CEX) is often used to assess deamidation or disulphide isoforms [2]. Enzyme linked immunosorbent assay (ELISA) are commonly used to analyse for host cell protein contaminations [3], and hydrophilic interaction liquid chromatography (HILIC) may be used to assess overall glycoprofiles [4]. These assays are numerous and time consuming in their entirety and many, such as CEX only indirectly measure the PQA of interest. Thus, while it may be able to determine product consistency, it may be unable to establish a firm connection between any specific attributes of interest and any process changes that have occurred.
Liquid chromatography mass spectrometry (LC-MS) is a highly accurate and sensitive platform routinely used in biopharmaceutical research and early development. The ability of LC-MS to thoroughly characterise a biologic with all its associated modifications through intact protein analysis, subunit analysis, peptide mapping and sequencing techniques has made the platform indispensable in modern discovery laboratories today [5]. Of particular interest here, modern peptide mapping techniques can confirm virtually the entire sequence of a protein or antibody based therapeutic and detect any post-translational and other modifications, their concentrations, and degrees of heterogeneity [6]. These same capabilities, in essence, are what are also required for monitoring biotherapeutics during manufacturing and QC, with the added benefit that LC-MS directly measures the attributes of interest. Historically, however, there has been much lower adoption in downstream laboratory environments due to real and perceived issues around specific workflow capabilities, cost, and ease-of-use [7].
More recently, an LC-MS peptide mapping based multiple attribute method (MAM) has been gaining traction within late-stage development and manufacturing laboratories [8]. The LC-MS peptide MAM workflow maximises efficiencies by combining the analysis of many PQAs into one assay. Similar to mass spectrometry based peptide mapping used in discovery, the LC-MS peptide MAM workflow can fully characterise a biotherapeutic in quality and quantity along with its associated modifications. This ability to replace many PQA assays, comprising many disparate technologies, with a single assay using a single technology makes LC-MS much more attractive and cost effective for downstream environments.
However, several challenges still exist within the MAM workflow itself before widespread adoption can be considered. Within the area of sample preparation, a peptide based MAM workflow requires intact proteins to be enzymatically digested into peptides prior to analysis [9]. Great emphasis must be placed on the reproducibility of this sample preparation protocol. In the areas of data acquisition and data analysis, the high resolving power of the mass spectrometer enables the detection of isotopically resolved peaks - meaning that every peak will have multiple smaller peaks that represent naturally occurring isotopes within the peptide. Additionally, the ionisation method used for the LC-MS MAM workflow typically produces multiply-charged ions during analysis, adding to the complexity of the spectra that are generated. Data processing software must routinely take these peak detection issues into account when identifying and quantifying peaks. New peaks that may arise due to any new impurities or contaminants entering the production process will further increase the spectral and chromatographic complexity of the data. These components need to be detected, but to date this has been one feature of the MAM workflow that has been missing, or difficult to routinely implement.
Because LC-MS is new to most late-stage development and manufacturing laboratories, users are generally unfamiliar with this analytical approach. Additionally, once implemented within a laboratory, LC-MS has typically required a level of operator expertise not normally associated with downstream laboratory environments. Thus, software for data acquisition and data processing must be intuitive and easy-to-learn without requiring extensive training or operators with advanced degrees. Hardware must be robust. The overall workflow must be extremely reproducible and reliable.
The following work describes implementation of a peptide-based LC-MS MAM workflow that addresses some of the concerns outlined above. A new peak detection feature provides the capability to monitor the purity of the biotherapeutic and flag potential new contaminants. The hardware is robust with acquisition software that is easy to use. Similarly, the processing software routines are integrated within one package such that both initial characterisation and routine monitoring are performed using the same features sets, simplifying learning and further streamlining analysis. These improvements are a firm step towards making the LC-MAM workflow more acceptable to downstream laboratory environments.
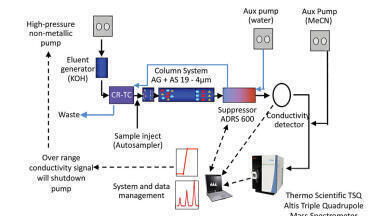
Sample Preparation: National Institute of Standards and Technology (NIST) monoclonal antibody reference standard material (10 mg/mL- NIST, MD, USA) was diluted to 1 µg/µL with denaturing buffer (7 M guanidine HCl, 100 mM Tris(hydroxymethyl)-aminomethane, pH 8.3- Sigma-Aldrich, MO, USA). To reduce the sample, 2 µL 500 mM Dithiothreitol (DTT) in denaturing buffer was added and the sample was mixed gently before incubating for 30 min at room temperature. The antibody was then alkylated by adding 4 µL 500 mM sodium iodoacetate in denaturing buffer and incubating in the dark at room temperature for 20 min. The remaining alkylating agent was quenched by adding 4 µL of 50 mM DTT in denaturing buffer. The final volume of sample was 110 µL. The antibody was then buffer transferred into 50 mM Tris HCl pH 7.9 using a BioSpin-6 column from Bio-Rad. Assuming 10-20% loss of antibody in the spin column, trypsin Gold stock solution (50 mM acetic acid, 1 µg/µL) at 1:10 enzyme:substrate ratio was added. The sample was vortexed briefly then allowed to incubate at 37°C for 30 min. The reaction was quenched by adding 1:10 (v/v) with 10% TFA. To test new peak detection, a spike in solution consisting of 20 peptides (PepCalMix, Sciex) was added to the sample at a concentration of 0.25 fmol/µl, such that 500 fmol of the spike in was injected on column. The sample was centrifuged and the liquid was transferred to a vial for injection.
Reversed Phase Liquid Chromatography: An ExionLC™ system, consisting of a Controller, two AD pumps, as well as AD autosampler, degasser and column oven was used for peptide separations (SCIEX, CA, USA). The column was a 150 x 2.1 mm Zorbax 300 SB-C18 1.8 µm 100 Å (Agilent, CA, USA). Mobile phase A, water with 0.1% formic acid, and mobile phase B, acetonitrile with 0.1% formic acid, was used at a flow rate of 0.25 ml/min. Wash solvent for the autosampler was 20/20/60 methanol/acetonitrile/IPA. Injection volume was 2-5 µL, and the column was kept at 50°C. The gradient method was as follows: 0 min, 1% B; 5 min, 1% B; 6 min, 10% B; 70 min, 35% B; 75 min, 90% B; 80 min, 90% B; 80.5 min, 1% B; 83.5 min, 10% B; 91.5 min, 45% B; 93.0 min, 90% B; 99 min, 90% B; 101 min, 1% B; 115 min, 1% B.
Mass Spectrometry: A Sciex X500B QTOF System with Turbo V source was used. LC-MS/MS data for initial sample characterisation was acquired using SWATH® data-independent acquisition (DIA). TOF MS data were acquired from m/z 300 to 1800, with an accumulation time of 250 ms and MS/MS fragment data were acquired from m/z 50 to 1800 using 25 windows at 50 ms per window. Accurate mass LC-MS data for routine data monitoring were acquired using TOF-MS only, acquiring data from m/z 300 to 1800 using an acquisition time of 1 second.
Software: Data Acquisition – Sciex OS Software was used for both the SWATH® acquisition for product characterisation as well as for routine TOF-MS analysis in the MAM assay. Data Processing – A Research Version of BioPharmaView™ Software was used for all aspects of data processing. This included initial characterisation of the antibody peptide digest and quantitation of all PQAs from the LC-MS/MS data, followed by monitoring, quantitation, purity testing, and reporting using the accurate mass TOF-MS data.
The first step in developing a peptide-based LC-MS MAM workflow is to fully characterise a reference standard to define attributes and set acceptance criteria. Typically, initial characterisation of the reference standard is accomplished through an acquisition strategy called data dependent acquisition (DDA); however, due to the stochastic nature of DDA, multiple rounds may be required in order to exhaustively characterise every component within the reference standard [10]. In this study, characterisation was performed using SWATH® acquisition – a data independent acquisition strategy (DIA). This technique collects data across the entire precursor mass range ensuring that data are generated from every detectable peptide, modification, impurity, or other analyte in the reference standard all within a single analysis [11]. The use of the SWATH acquisition workflow creates a complete and comprehensive archive where results can be interrogated in the future for new biological information. SWATH acquisition has the further advantage that it requires virtually no method development making it a simple and generic method for characterising biotherapeutics coming through the pipeline.
In this study the data for the reference standard was acquired and then processed using the BioPharmaView software package to identify and quantify all of the PQAs and other components within the reference sample. From this fully characterised data set, the calculations for the routine monitoring of all attributes of interest were then defined. Figure 1 depicts the setup for monitoring mannose 5 (Man5) on the Heavy Chain T25 peptide of the NIST antibody, a glycosylation attribute identified and characterised within the SWATH acquisition data. Here, the automated calculation of the percent of peptides containing Man5 glycosylation compared to the total glycosylated peptide pool is set in the software for simplified data analysis during routine monitoring. The calculation uses extracted ion chromatograms (XICs) of the high-resolution TOF MS data for multiple charge states of the Man5 glycosylated peptide, divided by the total XIC areas of the total glycopeptide pool, including the non-glycosylated peptide. Final calculated results are displayed on the left-side of the display. In this case 1.32% of the T25 peptide contains the Man5 glycosylation modification, as shown highlighted in blue. Thus, the amount of Man5 found in any test sample must fall within a pre-determined range around this reference amount (e.g., +/- 8%).
Once the reference standard is characterised and the targeted PQAs have been defined in the MAM processing method, this method can be deployed during routine monitoring. In routine sample monitoring fresh digests of both the reference standard and a test sample are analysed using high accuracy TOF-MS acquisition and processed against the developed criteria. To simplify the workflow, the data is processed using the same BioPharmaView processing software that was used for initial characterisation of the reference standard. Processing of the sample data in comparison to the known reference standard data yields an output indicating the number of unique peptides found in each sample, the number of known impurities detected, as well any newly detected peaks. Known impurities can be defined in the MAM method setup, and can include host cell proteins or sample preparation components that may be expected to be present, such as trypsin enzyme. Newly detected peaks that produce a unique signal in the test sample above a defined threshold will automatically be flagged and can be set to trigger sample failure.
In this study, TOF-MS data was acquired for the digested NIST antibody reference standard and for a test sample consisting of NIST standard with a peptide mix spiked in for new peak detection testing. As shown in Figure 2, the BioPharmaView software outputs the level detected for each PQA in the reference and test sample. Additionally, each attribute is annotated with either a green check to indicate passing criteria, a yellow triangle if levels fall within the marginal criteria, or a red circle if levels fall outside the set criteria. Overlays of the total ion chromatograms provide a visual output of the correspondence between the reference standard (blue trace) and test sample (pink trace, Figure 2B). Filtering for the Man5 glycosylated peptide attribute Figure 2A displays the accurate mass TOF MS extraction of the peptide (Figure 2B bottom) for both the standard (blue trace) and sample (pink trace). Newly detected peaks in the test sample are listed in the ‘Unmatched’ tab in the peptide results table, with m/z, charge state, retention time, and peak area noted. In this study 49 new peaks were found in the test sample, corresponding to the 20 peptides from the peptide spike-in mix and their multiple charge states.
An important last step of the MAM workflow is the ability to automatically generate a final report with all relevant information clearly and concisely summarised. Within the BioPharmaView software, automated reports can be generated for the MAM assay results, summarising sample PQA values compared to the reference standard, in addition to overall pass or fail grades. For example, as shown in Figure 3, report components include the target value and range for each defined PQA, as well as the calculations used to generate the results. This summary enables quick screening of the results to ensure the biotherapeutic sample passes defined specifications. In this case, the detection of a single pre-defined impurity of trypsin enzyme was found. Additionally, 49 unique peaks were found in the test sample, coming from the peptide spike mix, and the detection of these new peaks caused an overall ‘Fail’ grade for the biotherapeutic analysis.
LC-MS is relatively new in downstream biopharmaceutical laboratory environments and is currently viewed as a complementary technique. Before it can be seen as a disruptive/replacement technology, challenges must be overcome around cost, compliance, workflow gaps, and ease-of-use. The peptide-based accurate mass LC-MS MAM workflow outlined here addresses some of these challenges by providing a solution with new peak detection capabilities and software advancements. The new peak detection feature allows monitoring of PQAs while simultaneously detecting any new attributes, impurities, and contaminants that may occur. The acquisition software provides a simple approach for full characterisation and monitoring. The processing software streamlines data analysis with an all-in-one solution for initial PQA characterisation, routine monitoring, quantitation, purity testing and reporting. While there may still be hesitation in downstream laboratories around acceptance of LC-MS as a new analytical technique, and challenges around initial cost justifications, the overall benefits of the LC-MS MAM workflow to directly monitor more PQAs than other conventional characterisation methods, in addition to the current workflow improvements in ease-of-use/learning and new peak detection, may help accelerate acceptance by manufacturers and regulators alike.
1. Guidance for Industry, Q8(R2) Pharmaceutical Development, U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER), November 2009 ICH Revision 2: https://www.fda.gov/downloads/drugs/guidances/ucm073507.pdf
2. Chromatographic analysis of the acidic and basic species of recombinant monoclonal antibodies. Yi Du, Alison Walsh, Robin Ehrick, Wei Xu, Kimberly May and Hongcheng Liu. mAbs. 2012 4(5): 578-585 https://dx.doi.org/10.4161%2Fmabs.21328
3. Comparison of platform host cell protein ELISA to process-specific host cell protein ELISA. Feny Gunawan, Julie Nishihara, Peter Liu, Wendy Sandoval, Marty Vanderlaan, Heidi Zhang, Denise Krawitz. Biotechnol. Bioeng. 2017 Oct; http://dx.doi.org/10.1002/bit.26466
4. A rapid sample preparation method for mass spectrometric characterization of N-linked glycans. Ying Qing Yu, Martin Gilar, Jennifer Kaska, John C. Gebler. Rapid Comm. In Mass Spec. 2005 19(16): 2331-2336
5. Towards Sensitive and Accurate Analysis of Antibody Biotherapeutics by Liquid Chromatography Coupled with Mass Spectrometry. Bo An, Ming Zhang, Jun Qu. Drug Metabolism and Disposition. 2014: 42 (121): 1858 -1866 https://doi.org/10.1124/dmd.114.058917
6. Rapid comparison of a candidate biosimilar to an innovator monoclonal antibody with advanced liquid chromatography and mass spectrometry technologies. Hongwei Xie, Ashish Chakraborty, Joomi Ahn, Ying Qing Yu, Deepalakshmi P Dakshinamoorthy, Martin Gilar, Weibin Chen, St John Skilton, Jeffery R Mazzeo. mAbs 2010. 2(4) 379 – 394. 10.4161/mabs.2.4.11986
7. Characterizatio and QC of biopharmaceuticals by MS-based ‘multi-attribute’: advantages and challenges. Yun Zhang, Jingzhon Guo. 2017 9(6) 499-502. https://www.future-science.com/doi/pdf/10.4155/bio-2017-0004
8. Rogers, R.S., et. al., Development of a Quantitative Mass Spectrometry Multi-Attribute Method for Characterization, Quality Control Testing and Disposition of Biologics, mAbs, 7:5, 881-890: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4623056/pdf/kmab-07-05-1069454.pdf
9. LC-MS multi-attribute method for characterization of biologics. Journal of Applied Bioanalysis. 2017 3 (2) 21-25. http://dx.doi.org/10.17145/jab.17.003 (ISSN 2405-710X)
10. Targeted Data Extraction of the MS/MS Spectra Generated by Data-independent Acquisition: A New Concept for Consistent and Accurate Proteome Analysis. Ludovic C. Gillet, Pedro Navarro, Stephen Tate, Hannes Röst, Nathalie Selevsek, Lukas Reiter, Ron Bonner, and Ruedi Aebersold, Mol Cell Proteomics. 2012 Jun; 11(6): https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3433915/
11. Multi-laboratory assessment of reproducibility, qualitative and quantitative performance of SWATH-mass spectrometry. Ben Collins, Christie Hunter, Yansheng Liu, Birgit Schilling, George Rosenberger, Samuel Bader, Daniel Chan, Bradford W. Gibson, Anne Claude Gingras, Jason Held, Mio Hirayama-Kurogi, Guixue Hou, Christoph Krisp, Brett Larsen, Liang Lin, Siqi Liu, Mark Molloy, Robert Moritz, Sumio Ohtsuki, Ralph Schlapbach, Nathalie Selevsek, Stefani Thomas, Shin Chen Tzeng, Hui Zhang, and Ruedi Aebersold, Nature Communications. 2017 Jun; 8 (291



















-(1).jpg)