
Bioanalytical
Published over 12 years ago. See the latest and most current information on Bioanalytical.
Given its suitability for the analysis of volatiles, it might be thought that GC would not be associated with the study of bacteria. However, volatile organic compounds are being explored as biomarkers for early diagnosis of diseases including respiratory, gastrointestinal and urinary tract infections. For example, Clostridium difficile infection (CDI) a nosocomial-infection prevalent around the world, may be determined by a new,
novel approach employing a designer enzyme substrate to liberate a unique VOC profile. This novel approach using HS-SPME-GC-MS could be used alongside conventional methods for Cl. difficile detection, including toxin detection methods, which would allow any false negative results to be eliminated.
The presence of volatile organic compounds (VOCs) is ubiquitous in the world in which we live. Without any known activation we are able to detect a vast range of VOCs via our olfactory senses. The nose, with its inherent sensory array, coupled with our data processor (brain) detect VOCs as a vast range of smells and odours to which we then ascribe a range of subjective descriptors e.g. grassy, fruity, citrus, sweet, smoky, earthy and sulphury. Our inherent ability to detect a vast range of VOCs and ascribe some descriptor is limited only by our choice of vocabulary. In this context we are ‘exposed’ to VOCs in a variety of contexts. For example, in the domestic environment we encounter VOCs as odours from a range of sources including many household products e.g. washing-up liquid (lemon / pine); cooking processes e.g. food preparation (aromatic / herbal); and, personal hygiene products e.g. soap and anti-perspirant deodorant (herbal / pine). In addition, we also readily identify a range of odours that we find repugnant i.e. malodour. In this situation we might encounter such smells associated with our daily ablutions (e.g. so-called toilet smells emanating from our gastrointestinal tract) as well as smells associated with our own hygiene e.g. halitosis and sweat (foot and/or underarm odour). [Note: Obviously we also encounter VOCs in many other contexts, for example, as environmental pollutants (i.e. vehicle exhaust fumes, industrial processes and atmospheric contamination). However, in the context of this article these have been ignored.] A range of VOCs and their associated subjective descriptor are shown in Figure 1. In fact the art of smell (because that is what it is) requires some ‘training’; ultimately we cannot be sure that we all associate the same VOC with the specific odour (even if we apply the same subjective descriptor). As we know we normally remove the subjectivity of people from the determination of VOCs and use instrumental approaches based on gas chromatography (GC) or gas sensors. The exception to this is the combined GC-olfactory detector which combines the instrumental and human interface to detect odour and its concentration.
In the health-related research focus of this article VOC determination has been done in a range of contexts [1-12]. For example, the detection of VOCs in exhaled air as biomarkers of diseases including lung cancer [1-6], gastrointestinal and liver disease [7]; as well as detection of VOCs emanating from urine (e.g. as an approach for assessing tuberculosis patients [8]) and faeces (e.g. diagnosis of gastrointestinal disease) [9,10]. In addition some excellent reviews are available including a review of clinical applications of VOC analysis for detecting infectious diseases, specifically respiratory, gastrointestinal and urinary tract infections [11]; in addition, an extensive review of the VOCs emanating from bacteria has been published [12].
A range of different techniques have been applied for the determination of the VOCs including thermal desorption gas chromatography mass spectrometry [8], solid-phase microextraction gas chromatography mass spectrometry [2, 5, 7, 9, 13], headspace-GC-MS [8, 10], proton transfer reaction mass spectrometry [14], selected ion flow tube mass spectrometry [15], ion mobility spectrometry [15] as well as electronic noses [1, 3]. In our research we have investigated a range of different VOCs associated with specific bacteria, specifically Cl. difficile [13].
Background to Cl. difficile
Clostridium difficile, originally named as Bacillus diffficilis [16] was first described in 1935. However, it was not for another 40 years that the importance of Cl. difficile was determined [17]. Cl. difficile is a Gram-positive, spore forming enteric anaerobic pathogen associated with pseudomembraneous colitis and is the main infectious cause of nosocomial diarrohea induced by antibiotic treatment. Cl. difficile flourishes in the human bowel, after antibiotic treatment, as a direct result of the modification of the normal balance of intestinal flora. Two exotoxins (enterotoxin A and cytotoxin B) are produced by pathogenic strains which lead to Cl. difficile infection (CDI). One of the main clinical symptoms of CDI is diarrhoeal stools. These have been defined as those that take the shape of the container [18] or type 7 on the Bristol Stool form scale [19].
Current methodology to diagnose Cl. difficile-associated disease
A range of approaches have been investigated and developed to allow diagnosis of CDI. These include non-microbiological methods (e.g. clinical assessment, endoscopy, faecal leukocytes and lactoferrin), detection of Cl. difficile products (e.g. glutamate dehydrogenase, volatile fatty acids and toxins), detection of Cl. difficile genes (e.g. 16s rRNA, toxin genes) and isolation and typing of Cl. difficile (e.g. culture and identification, typing and toxin testing, and antibiotic sensitivity testing) [18]. The most common methods of routine testing in laboratory diagnosis are those related to the detection of Cl. difficile products, and specifically toxins, in stool samples. Currently the most common testing procedure is immunoassay [18, 20, 21]; this is due to its relatively low cost, rapid turnaround, and high specificity [22]. Enzyme immunoassay does still suffer from low sensitivity leading to false negative results [22]. As a consequence isolation of Cl. difficile by culture (and subsequent demonstration of toxin production) is regarded as the ‘gold’ standard in spite of the delayed time period to obtain a result (48 hours) [23].
The role of gas chromatography for identification of Cl. difficile
One approach that was proposed and investigated in the 1980s was whether it was possible to utilise the separation potential and capability of gas chromatography (GC) to detect the volatile metabolites which may be indicative of Cl. difficile. It is interesting to note that most studies at that time used packed column GC which led to inferior separation compared to modern fused silica column technology (which were actually developed in 1979)[24]. Several approaches were adopted to investigate the suitability of GC for Cl. difficile identification based on VOC determination and these were: broth media; bacterial colonies from agar plates; and, direct stool sample analyses.
Moss and Nunez-Montiel [25] used the new fused silica capillary column to separate short-chain fatty acids derived from Cl. difficile. The bacteria were grown in a basal broth medium (trypticase yeast extract-salts broth) for 5 days prior to liquid-liquid extraction, of the spent growth medium, with diethyl ether. The acids present were then derivatised to form their trifluoroacetyl butyl esters. Extracts were analysed by both GC-FID and GC-MS. The results from a spent broth culture medium from Cl. difficile strain CDC A567 were identified to contain acetic, propionic, isobutyric, butyric, isovaleric and isocaproic acids. In addition, other major compounds identified were phenylacetic acid and hydrocinnamic acid, together with moderate amounts of indole-acetic acid and 2-ketobutryric acid as well as a small amount of p-hydroxyphenylacetic acid. Two unidentified sulphur containing compounds were also eluted from the column but not identified. In all cases identification was obtained by confirmation of both EI and CI mass spectral databases with authentic standards.
The same group [26] used a norleucine-tyrosine (NT) broth for the identification of Cl. difficile on the basis of caproic acid and p-cresol evolution. In a similar approach, liquid-liquid extraction was used to extract the compounds from the broth, using either ether or chloroform, and analysing the resultant extract with packed GC-TCD. A total of 120 strains of Cl. difficile were investigated including 2 stock strains and a further 118 strains isolated from faecal samples. All strains investigated produced caproic acid and p-cresol in the NT broth within 24-48 hours of incubation. Other compounds identified included acetic, isobutyric, butyric, isovaleric, valeric and isocaproic acids. The authors proposed that the identification of both caproic acid and p-cresol from NT broths was sufficiently reliable for identification of Cl. difficile.
Further developments by the group [27] led them to explore a range of amino acid broths as culture media for Cl. difficile (and specifically strain CDC A-567). Cl. difficile was cultured in trypticase yeast salt broth supplemented with L-leucine, L-norleucine, L-isoleucine, L-tyrosine or L-trypotophan. The spent medium was then extracted using either chloroform or ether. The extracts were then derivatised with either trichloroethanol, heptafluorobutryic anhydride or heptafluorobutryic anhydride ethanol and analysed using packed GC with a frequency pulsed electron capture detector (provides extra sensitivity for halogenated compounds). It was found that isocaproic acid was produced in relatively high concentration irrespective of the growth medium used. It was proposed that this approach could form the basis of a rapid detection system for isocaproic acid in Cl. difficile in stool samples. It was noted however that reliance on detection of isocaproic acid may be problematic as Cl. bifermentans, which also produces isocaproic acid, can also be present in stool samples. The authors suggest that the identification of compounds other than carboxylic acids should provide the basis to differentiate between Cl. bifermentans and Cl. difficile.
A similar approach was adopted by Johnson et al. [28] in which stool samples from 746 patients, from two different medical centres, were tested for the presence of Cl. difficile. Stool samples were cultured for 48 hours in Beckton Dickinson supplemental peptone broth to which cefoxitin was added. A 1 mL aliquot of the broth was then removed and subjected to methylation in acid solution at 56°C for 30 min. This sample was then extracted in to chloroform and analysed by packed GC-FID. In the presence of Cl. difficile four distinctive peaks could be identified: an unresolved peak immediately prior to isovaleric acid; phenylacetic acid; isocaproic acid; and, hydrocinnamic acid. It was noted that as phenylacetic acid and hydrocinnamic acid are not volatile acids their identification would not be possible except without methylation of the extract. The authors proposed that this approach eliminated the need to sub-culture for tests requiring a pure isolate. They proposed that the success of the method in detecting four fatty acids, resulting from the metabolic end product of Cl. difficile, was dependent upon the precise preparation of the sample; specifically the use of freshly thawed cefoxitin. Failure to adhere to the procedure would result in false positive results being obtained.
Sivsammye and Sims [29] adopted the idea of Levett and Phillips (1985) by using broth supplemented with p-hydroxyphenylacetic acid for the presumptive identification of Cl. difficile in a more rapid timescale i.e. 18 hours. p-Hydroxyphenylacetic acid was targeted for this broth as it has been shown that Cl. difficile decarboxylates it to p-cresol [30, 31]. A total of 282 organisms was tested including 47 stock strains of Cl. difficile, 180 test organisms isolated on brain heart infusion agar-cefoxitin from 80 patients, Cl. difficile ATCC 43593 and ATCC 43594, as well as 53 negative control species, were analysed. The broth used was pre-reduced, anaerobically sterilised (PRAS) peptone yeast glucose (PYG) broth supplemented with p-hydroxyphenylacetic acid for a one step identification of Cl. difficile based on the analysis of p-cresol by packed GC-FID. It was found that all 49 stock and reference strains of Cl. difficile and 19 organisms confirmed as Cl. difficile produced p-cresol. It was noted that Cl. difficile did not produce p-cresol in the broth (PYG) without the addition of p-hydroxyphenylacetic acid; also, p-cresol was not detected in uninoculated broth or broth with p-hydroxyphenylacetic acid.
In order to speed up the identification process of Cl. difficile from faeces an alternative approach was adopted by Levett and Phillips [32]. The identification of Cl. difficile from faeces via selective media and procedures involving the isolation of pure cultures and biochemical testing can take up to 5 days. Therefore any approach that can speed up the early diagnosis of Cl. difficile is beneficial in the diagnostic laboratory. In their research they obtained 190 stool samples, from hospitals around the UK, from patients with diarrhoea thought to have resulted from the presence of Cl. difficile. All samples were cultured on modified cycloserine cefoxitin fructose agar (CCFA) medium plates and incubated for 48 h at 37°C. For the packed GC-FID analysis a plug of agar was removed from an area of the plate containing growth of suspected Cl. difficile. To this sample was added 1 drop of water which was left for 10 min at room temperature. Then, 1 µL of the aqueous extract was analysed by GC-FID. Analysis of pure cultures of Cl. difficile on the modified CCFA medium, without antibiotics, identified isocaproic acid, caproic acid and p-cresol. In addition, analysis of Cl. bifermentans, Cl. sordellii and Cl. sporogenes identified isocaproic acid, caproic acid, gamma-amino butyric acid and δ-amino valeric acid. Isocaproic acid, caproic acid and p-cresol were not detected in uninoculated CCFA medium; these compounds were also not identified in cultures of Cl. butyricum, Cl. glycolicum, Cl. innocuum or Cl. paraputrificum. However, it was possible to identify isocaproic acid and p-cresol (but not caproic acid) from Cl. scatologenes NCTC 9800. Cl. difficile was isolated in 35% of all stool samples analysed i.e. 66 samples. A characteristic pattern of peaks was identified for the presence of isocaproic acid, caproic acid and p-cresol in 66 of the stool samples; all of these samples produced a growth of Cl. difficile. It was noted that no other inoculated plates contained all of these metabolites. The authors proposed that the use of a plug of modified CCFA medium facilitated definitive identification of Cl. difficile within 24-48 hours.
An approach to use GC as a screening tool for toxigenic Cl. difficile in diarrhoeal stools was used [33]. In this case a portion of the stool sample (1.5 mL) was mixed with phosphate-buffered saline (PBS) and, after acidification, extracted with ether. The resultant extract was then analysed by packed GC-FID. Fatty acid compounds were identified by comparing their retention times with those of known standards. A total of 154 stool samples were analysed; of these, 129 samples produced no significant peak (< 1.2 cm) for isocaproic acid and were also found to be toxin-negative. The authors concluded that the lack of a ‘significant’ isocaproic acid peak was a rapid screening test for excluding Cl. difficile infection; moreover positive results (i.e. a ‘significant’ isocaproic acid peak identified) must then be checked by toxin testing and culture for the presence of Cl. difficile (and its associated toxin).
In direct contrast however, Levett [34] reported that the GC analysis of fatty acids or p-cresol in faeces was not satisfactory as a screening test for the presence of Cl. difficile. In this work 110 stool samples were obtained from around the UK from patients suspected of suffering from CDI. A 50% (w/v) suspension of faecal samples was mixed with PBS, and after acidification, extracted with diethyl ether, and analysed by packed GC-FID. The author reported no association between the presence of Cl. difficile or cytotoxin and acetic, propionic, isobutyric, butyric or valeric acids. However, significant associations were found between Cl. difficile and the presence of isovaleric acid (isovaleric acid detected in 85% of stool samples), Cl. difficile and isocaproic acid (isocaproic acid detected in 41% of stool samples) and Cl. difficile and p-cresol (p-cresol detected in 52% of stool samples). However, no correlation was found between Cl. difficile and the combined presence of isovaleric acid, isocaproic acid and p-cresol. The author concluded that the high rate of false negatives associated with the detection of isocaproic acid and p-cresol coupled with the fact that other organisms (Cl. bifermentans, Cl. sordellii and Cl. sporogenes) can produce isocaproic acid makes the GC approach unreliable as a screening test.
However, further support for the use of isocaproic acid as a marker for the screening of CDI was reported [35]. In this study [35] 90 stool samples were investigated using packed GC-FID and the results compared with both culture on a selective medium and cytotoxin assay in tissue culture. Faecal samples were extracted in either acidified ether or acidified PBS in ether. Identification of eluted compounds was done using known standards and their retention times. Using a combined determination of both isocaproic acid and butyric acid (butyric acid was chosen arbitrarily as representative of volatile fatty acids as it was present in almost all samples; 8 samples did not contain butyric acid) the authors were able to identify three categories: positive, negative and indeterminate. A positive result was indicated by isocaproic acid having a peak height > 0.5 cm; for samples with a peak height ≤ 0.5 cm were also considered positive provided the peak height for butyric acid was ≤ 5 cm. Negative samples were characterised by having either no signal for isocaproic acid or a signal ≤ 0.5 cm and butyric acid > 5 cm. Samples were classed as indeterminate if they had no discernible peak for isocaproic acid and a peak for butyric acid of ≤ 5 cm. By excluding the indeterminate group it was possible to use the predictive capability of GC to confirm 87% of positive cases and 85% of negative cases compared to culture on a selective medium (cycloserine-cefoxitin-fructose agar) and 71% of positive cases and 95% of negative cases compared to cytotoxin assay. The authors identified that the number of false positives was relatively small (11%) when compared to the culture method; however, the percentage of false negatives was large (41%). In comparison with the cytotoxin assay however the percentage of false negatives was fewer (27%). They concluded that their semi-quantitative approach using isocaproic acid and butyric acid provided a rapid provisional diagnosis with a high predictive value in at least 2/3rds of cases. They suggested that their GC approach could be helpful to decide whether to start prompt and appropriate treatment while obtaining definitive diagnosis of Cl. difficile associated disease by alternative approaches e.g. cytotoxin assay, along with further decisions about patient treatment.
The use of GC to identify the VOC profile from Cl. difficile in a clinical environment was however rapidly rejected as a suitable way forward [18, 32] due to the contradictory results reported [32]. Nevertheless development in GC technology including the ability to effectively separate multi-component mixtures and the use of the mass spectrometer, as the detector of choice, has meant that some research has re-emerged in recent years [37, 38]. While the paper by De Preter et al. [38] is concerned with the optimisation of the purge and trap system for screening faecal samples; data is provided however on the identities of VOCs from 11 stool samples from healthy volunteers. The analysis of the faecal samples identified 135 different VOCs of which 22 were found in all volunteers. In contrast, Garner et al. [37] investigated, using SPME-GC-MS, VOCs profiles from both healthy donors and those patients with gastrointestinal disease (including Cl. difficile). In total 111 stool samples were obtained of which 22 were from patients suffering from Cl. difficile. The research identified 297 VOCs across all samples of which the following were present in all samples: ethanoic acid, butanoic acid, pentanoic acid, benzaldehyde, ethanol, carbon disulphide, dimethyldisulphide, acetone, 2-butanone, 2,3-butanedione, 6-methyl-5-hepten-2-one, indole and 4-methylphenol. Specifically 145 VOCs were found in stool samples from patients diagnosed with Cl. difficile. Discriminant analysis was done on selected VOCs (32 specific and identified compounds) from the stool samples of asymptomatic volunteers, patients with either Campylobacter jejuni, Cl. difficile or ulcerative colitis, and was found to produce clustering of cases into 4 distinct groups. The authors indicated that SPME-GC-MS is an effective approach for the rapid qualitative analysis of the VOC profile of stool samples. They also postulated that it should be possible to differentiate between diseases by identification of a small number of compounds.
Development of a gas
chromatography based approach for identification of Cl. difficile
A new approach was therefore required if GC was to have any potential for early diagnosis of Cl. difficile. The new approach sought to utilise previous work [30, 31] that had indicated that Cl. difficile decarboxylates p-hydroxyphenylacetic acid to p-cresol. The basis of the developed approach was to add a designer enzyme substrate to the sample that would be liberate a unique VOC characteristic of Cl. difficile only. The selected designer enzyme substrate was 3-fluoro-4-hydroxyphenylacetic acid (FHPAA) which would decarboxylate in the presence of Cl. difficile to liberate the VOC 2-fluoro-4-methylphenol.
Experimental
Chemicals and reagents
Isobutyric acid (99%), butyric acid (≥ 99%), isocaproic acid (99%), caproic acid (99%), p-cresol (99%) and 3-fluoro-4-hydroxyphenylacetic acid (98%) as well as D-cycloserine, amphotericin and cefoxitin sodium salt were purchased from Sigma-Aldrich (Poole, UK). 2-Fluoro-4-methylphenol (98%) was obtained from Alfa Aesar (Morecambe, UK). All solvents were of analytical reagent grade and purchased from Fisher Scientific (Loughborough, UK). Cooked meat granules and CCEY agar (cycloserine cefoxitin egg yolk agar or Brazier’s agar) were obtained from BioConnections (Wetherby, UK). Brain heart infusion (BHI) broth and Columbia blood agar were purchased from Oxoid (Basingstoke, UK) and Taurocholic acid, sodium salt (≥ 90%) from Calbiochem (Nottingham, UK). The chromogenic agar chrom ID Cl. difficile used for determining method sensitivity was obtained from bioMérieux (Marcy l’Etoile, France). SPME fibres (85 µm polyacrylate (PA)) for extracting bacterial VOCs were purchased from Supelco Corp. (Bellefonte, PA, USA). All fibres were conditioned in the GC injection port prior to use as directed by manufacturer’s guidelines. All fibres were used with a manual holder.
Instrumentation
Gas chromatography/mass spectrometry (GC/MS) analysis was performed on a Trace GC Ultra and Polaris Q ion trap mass spectrometer (Thermo Scientific, Hemel Hempstead, UK) with Xcaliber 1.4 SR1 software. Separation of VOCs was carried out using a 30 m x 0.25 mm ID x 0.25 µm VF-waxMS capillary column (Varian, Agilent Technologies, Stockport, UK). The temperature program used was: 50°C held for 2 min then increased at a rate of 10°C/min to 220°C with a final 2 min hold. The split-splitless injection port was held at 230°C for desorption of volatiles in split mode at a split ratio of 1:10. Helium was used as the carrier gas at a constant flow rate of 1.0 mL/min. MS parameters were as follows: full-scan mode with scan range 50 – 650 amu at a rate of 0.58 scan/s. The ion source temperature was 250°C with an ionising energy of 70 eV and a mass transfer line of 250°C.
Identification of VOCs was achieved using the National Institute of Standards and Technology (NIST) reference library (NIST Mass spectral library, version 2.0a, 2001) as well as the comparison of the retention times and mass spectra of authentic standards. In addition, a dedicated mass spectral library was built in-house using mass spectra of authentic compounds to confirm the identity of detected VOCs.
Microbiology
Cl. difficile ribotypes R-015, R-106 and R-064 were obtained from the Microbiology Department, Freeman Hospital, Newcastle upon Tyne. All bacteria were sub-cultured on Columbia blood agar with 5 % defibrinated horse blood and incubated at 37°C under anaerobic conditions. Stool samples were clinical samples and were obtained from the Microbiology Department at the Freeman Hospital where they were confirmed as Cl. difficile culture positive, toxin positive or Cl. difficile culture positive, toxin negative or Cl. difficile culture negative, toxin negative. All stool samples were tested for GDH (glutamate dehydrogenase) which is an enzyme produced by Cl. difficile. To culture Cl. difficile, samples were first subject to alcohol shock. This included emulsifying a small volume of sample with an equal volume of 96% ethanol and left at room temperature for 30 minutes. Then, 50 µL of this suspension was inoculated onto CCEY agar and plates were incubated anaerobically at 37°C for 48 hours. Growth of Cl. difficile was identified by colonial appearance, fluorescence under UV light and MALDI-TOF- mass spectrometry (Bruker, Coventry, UK). To test if samples were Cl. difficile toxin positive or negative, samples were put through the VIDAS toxin A/B detection system (bioMérieux, Marcy l’Etoile, France). This involved sub-culturing Cl. difficile into a cooked meat broth and incubating for 48 hours. An aliquot of this broth was then centrifuged at 13,000 rpm for 5 minutes. The supernatant was then tested with the VIDAS toxin A/B detection system.
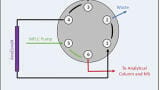
HS-SPME GC/MS procedure
Bacterial volatile organic compounds were extracted from the headspace of samples and concentrated via SPME before desorption in the hot GC injection port. All samples were held at 37°C in a water bath for 30 minutes prior to VOC extraction and kept at this temperature throughout sampling. A fused-silica SPME fibre with PA coating pierced the PTFE septum and was exposed in the headspace of the vial for 10 minutes. All fibres were conditioned according to manufacturer’s guidelines prior to use. Immediately after VOC extraction the SPME fibre was exposed in the hot GC injection port for 2 minutes for desorption of bacterial VOCs.
Results and Discussion
Identification and quantification of bacterial VOCs
Calibration graphs of the VOCs p-cresol, 2-fluoro-4-methylphenol and the fatty acids isobutyric acid, butyric acid, isocaproic acid and caproic acid were prepared by spiking VOC standards of known concentration into 10 mL cooked meat broth to which 2.5 g/L sodium taurocholate, 250 µg/mL D-cycloserine, 8 µg/mL cefoxitin and 4 µg/mL amphotericin were added, followed by incubation of spiked blank at 37°C and subsequent extraction of VOCs. HS-SPME procedure and GC/MS parameters were consistent with those used for bacterial VOC analysis. VOCs were quantified using external calibration and the values for limit of detection (LOD) and limit of quantification (LOQ) were determined as the peak area 3 times the signal to noise ratio and 10 times the signal to noise ratio, respectively. The results are shown in Table 1.
Application of method to stool samples
The developed method for analysis of stool samples was as follows: all samples were subjected to alcohol shock, centrifuged at 13,000 rpm for 5 minutes, ethanol removed and solid inoculated into 10 ml cooked meat broth. The broth contained 250 µg/mL D-cycloserine and 8 µg/mL cefoxitin, 4 µg/mL amphotericin, 100 µg/mL FHPAA and 2.5 g/L sodium taurocholate. The method was applied to 100 stool samples in a blind study with quantification of VOCs. A 10 mL blank cooked meat broth was analysed during every day of sampling. Post (HS-SPME-GC/MS) analysis the stool samples were confirmed as 60 culture positive, toxin positive; 17 culture positive, toxin negative and 23 culture negative, toxin negative. Typical chromatograms, obtained by HS-SPME-GC/MS of stool samples, for Cl. difficile culture +ve and toxin +ve; Cl. difficile culture +ve and toxin –ve; and, Cl. difficile culture –ve and toxin –ve are shown in Figure 2. Table 2 summarises the concentration of the six VOCs per stool sample while the statistical analysis of the entire sample dataset is shown in Table 3. It is concluded that 2-fluoro-4-methylphenol provides a unique identifier between Cl. difficile positive and Cl. difficile negative samples. It is not possible to differentiate between Cl. difficile positive toxic positive and toxic negative. No other fatty acid or p-cresol offers any form of selectivity to differentiate between Cl. difficile positive and negative samples.
Conclusions
The developed method allowed confirmation of the presence of Cl. difficile with very high specificity (100%) after 18 h. This innovative approach of exploiting novel enzyme substrates that release unusual VOCs that are not normally found in bacterial cultures, may find application in the detection of other bacterial pathogens in clinical or food microbiology.
Acknowledgements The financial support of bioMérieux SA is gratefully acknowledged. High resolution mass spectrometry was acquired at the EPSRC UK National Mass Spectrometry Facility at Swansea University. In addition, technical support from Mr E. Ludkin and Mr Gary Noble is also gratefully acknowledged.
References
[1] C. Belda-Iniesta, J. de Castro Carpeno, J.A. Carrasco, V. Moreno, E. Casado Saenz, J. Feliu, M. Sereno, F. Garcia Rio, J. Barriuso, M. Gonzalez Baron, Clin. Transl. Oncol., 9 (2007) 364.
[2] B. Buszewski, M. Kesy, T. Ligor, A. Amann, Biomed. Chromatogr., 21 (2007) 553.
[3] I. Horvath, Z. Lazar, N. Gyulai, M. Kollai, G. Losoncsy, Eur. Respir. J., 34 (2009) 261.
[4] H.P. Chan, C. Lewis, P.S. Thomas, Lung Cancer 63 (2009) 164.
[5] J. Rudnicka, T. Kowalkowski, T. Ligor, B. Buszewski, J. Chromatogra. B 879 (2011) 3360.
[6] M. Hakim, Y.Y. Broza, O. Barash, N. Peled, M. Phillips, A. Amann, H. Haick, Chem. Rev. 112 (2012) 5949.
[7] C.S.J. Probert, I. Ahmed, T. Khalid, E. Johnson, S. Smth and N. Ratcliffe, J. Gastrointestin. Liver Dis., 18 (2009) 337.
[8] M. Phillips, V. Basa-Dalay, G. Bothamley, R.N. Cataneo, P.K. Lam, M.P.R. Natividad, P. Schmitt, J. Wai, Tuberculosis 90 (2010) 145.
[9] C.E. Garner, S. Smith, B. de lacy Costello, P. White, R. Spencer, C.S.J. Probert, N.M. Ratcliffe, FASEB 21 (2007) 1675.
[10] K.M. Banday, K.K. Pasikanti, E.C.Y. Chan, R. Singla, K.V.S. Rao, V.S. Chauhan, R.K. Nanda, Anal. Chem., 83 (2011) 5526.
[11] S. Sethi, R.Nanda, T. Chakraborty, Clin. Microbiol. Rev., 26 (2013) 462.
[12] S. Schulz, J.S. Dickschat, Nat. Prod. Rev., 24 (2007) 814.
[13] E. Tait, K.A. Hill, J.D. Perry, S.P. Stanforth, J.R. Dean, J. Appl. Microbiol. Doi: 10.1111/jam.12418.
[14] D. Mayr, R. Margein, E. Klingsbichel, E. Hartungen, D. Jenewein, F. Schinner, T.D. Mark, Appl. Env. Microbiol. 69 (2003) 4697.
[15] J. Dummer, M. Storer, M. Swanney, M. McEwan, A. Scott-Thomas, S. Bhandari, S. Chambers, R. Dweik, M. Epton, Trends Anal. Chem., 30 (2011) 960.
[16] I.C. Hall, E. O’Toole, Am. J. Dis. Child, 49 (1935) 390.
[17] D.E. Voth, J.D. Ballard, Clin. Microbiol. Rev. 18(2) (2005) 247.
[18] A. Berrington, S.P. Borriello, J. Brazier, G. Duckworth, K. Foster, R. Freeman, F.K. Gould, J. Henderson, V. Hollyoak, R. Spencer, J. Taylor, M. Wilcox, J. Hospital Infection, 56 (2004) 1.
[19] Department of Health and Health Protection Agency. Cl. difficile infection: How to deal with the problem. (287860 / Cl. difficile infection), 2009.
[20] J.D. Perry, K. Asir, D. Halimi, S. Orenga, J. Dale, M. Payne, R. Carlton, J. Evans, F.K. Gould, J. Clin. Microbiol., 48(11) (2010) 3852.
[21] J. Swindells, N. Brenwald, N. Reading, B. Oppenheim, J. Clin. Microbiol., 48(2) (2010) 606.
[22] D.M. Cardona, K.H. Rand, J. Clin. Microbiol., 46(11) (2008) 3686.
[23] T.D. Wilkins, D.M. Lyerly, J. Clin. Microbiol., 41(2) (2003) 531.
[24] R.D. Dandeneau, E.H. Zerenner. J. High Resolut. Chromatogr., 2 (1979) 351.
[25] C.W. Moss, O.L. Nunez-Montiel, J. Clin. Microbiol., 15(2) (1982) 308.
[26] O.L. Nunez-Montiel, F.S. Thompson, V.R. Dowell, Jr., J. Clin. Microbiol., 17(2) (1983) 382.
[27] J.B. Brooks, O.L. Nunez-Montiel, B.J. Wycoff, C.W. Moss, J. Clin. Microbiol., 20(3) (1984) 539.
[28] L.L. Johnson, L.V. McFarland, P. Dearing, V. Raisys, F.D. Schoenknecht, J. Clin. Microbiol., 27(10) (1989) 2218.
[29] G. Sivsammye, H.V. Sims, J. Clin. Microbiol., 28(8) (1990) 1851.
[30] S. Hafiz, C.L. Oakley, J. Med. Microbiol., 9 (1976) 129.
[31] L. D’Ari, H.A. Barker, Arch. Microbiol., 143 (1985) 311.
[32] P.N. Levett, K.D. Philips, J. Clin. Pathol., 38 (1985) 82.
[33] F. Perersack, M. Labbe, C. Nonhoff, E. Schoutens, J. Clin. Pathol., 36 (1983) 1233.
[34] P.N. Levett, J. Clin. Pathol., 37 (1984) 117.
[35] P. Gianfrilli, A. Pantosti, I. Luzzo, J. Clin. Pathol., 38 (1985) 690.
[36] J.S. Brazier, J. Antimicrobial Chemotherapy, 41, Suppl. C (1998) 29.
[37] C.E. Garner, S. Smith, B. de Lacy Costello, P. White, R. Spencer, C.S.J. Probert, N.M. Ratcliffe, FASEB J., 21 (2007) 1675.
[38] V. De Preter, G. Van Staeyen, D. Esser, P. Rutgeerts, K. Verbeke, J. Chromatogr., A, 1216 (2009) 1476.





















-(1).jpg)