
Columns (LC)
Published over 13 years ago. See the latest and most current information on Columns (LC).
Ionic interactions are important in determining selectivity in HILIC separations. Knowing how to utilise these interactions in order to selectively change retention of analytes or interfering peaks in the chromatogram is a valuable tool when developing HILIC methods. By changing the ionisation state of the analytes and buffer strength of the mobile phase, retention of electrostatically repelled analytes can be increased or decreased depending on the extent to which the stationary phase surface repulsion is ‘shielded’ by the buffer salt.
Introduction
Hydrophilic Interaction Chromatography (HILIC) is a powerful technique for the separation of polar compounds. HILIC has emerged as the second most used HPLC technique after reversed phase chromatography. This growth has been all the more remarkable since the retention mechanism is still being debated and it is only just starting to be incorporated in University curricula.
There are numerous HILIC stationary phases with different bonded phase chemistries, most of which carry ionic charges, either deliberately incorporated during synthesis or from residual silanol groups. There is a general consensus that in HILIC a buffer salt needs to be used in order to control the ionic interactions between the analytes and the stationary phase. This is true also for nominally neutral stationary phases. Polar partitioning is the main retention mechanism in HILIC but the presence of ionic interactions can have a tremendous impact on retention. It is this mixed mode separation aspect which was explored in the study described in this paper.
Numerous papers have been published examining the differences between HILIC stationary phases and the effects on retention from altering chromatographic conditions. There are four distinct classes of HILIC stationary phases, neutral, anionic (silica), cationic (amine containing phases) and zwitterionic1,2.
A charged functional group on a column's surface has an order of magnitude greater free energy of interaction with charged analytes than that of an uncharged stationary phase (Table 1). These electrostatic interactions provide the possibility of changing retention time and controlling selectivity by altering pH and/or buffer strength. The strength of ionic interactions require, however, the addition of high concentrations of salt in order to overcome the electrostatic interactions between charged (anionic or cationic) stationary phases and charged analytes to promote reasonable retention.
Truly zwitterionic HILIC stationary phases also provide sites for such electrostatic interactions, but at a much lower magnitude, due to close proximity of ion and counter ion in balanced proportions within their functional groups. In the ZIC-HILIC column the distal charge, the sulphonate, will dominate the interaction and the phase will behave as a net cation exchanger1,3-4 at buffer concentrations low enough to expose analytes to this charge. The possibility of changing column selectivity is a very effective means of improving resolution and in HILIC (especially with zwitterionic phases) it is more easily utilised than in reversed phase chromatography. By changing the buffer strength or pH similar dramatic effects on selectivity can be achieved as when adding ion-pairing agents in reversed phase. This is of course quite complicated unless the charge state of the stationary phase is pH independent. It was therefore sought to demonstrate how compounds can be selectively moved to avoid co-elution with other similar compounds by simply adjusting pH and buffer strength.
Experimental
Preparation of a stock solution of buffer at two different pH simplified solvent preparation. Listed buffer concentrations in each experiment refer to final total concentrations. Ammonium formate at pH 3 was prepared using ammonium formate and adjusted with formic acid. The ammonium formate pH 6.5 was used without pH adjustment. A SeQuant® ZIC®-HILIC column, 150 x 2.1mm (5μm, 200 Å) was operated at a flow rate of 0.4mL/min, with an eluent of 70% ACN and 30% buffer (v/v) for all experiments. The chromatographic separations were carried out isocratically on a Shimadzu LC 10 system with column oven set at 30°C. The sample injected was 5µl with 150µg/ml of each compound dissolved in the eluent. Due to the analytes low UV absorbance detection was with a Sedere Sedex 85 ELSD equipped with a low flow nebuliser cell.
Three analytes were chosen for this study. Glutamine (Gln), which is a zwitterionic amino acid, with a hydrophilic side chain containing an amide functionality. Glutamine has two pKa values 2.2 and 9.1 thus exhibiting a stable state of ionisation in the studied pH range. Glutamic acid (Glu), which is an acidic amino acid with a carboxylic side chain has pKa values of 2.2, 4.2 and 9.7. α-Ketoglutaric acid, is a dicarboxylic acid with pKa values of 2.4 and 4.4.
Results
Acidic pH & low buffer strength
At pH 3.0 and 3mM buffer, the sidechain carboxyls (pKa ~4.2) will be principally protonated, and thus act as neutral polar groups, whereas the alpha carboxyls of Glu and Gln (pKa 2.2) and α-ketoglutarate (pKa 2.4) will be largely ionic and repelled by the negatively charged sulfonates of the column. In the amino acids Glu and Gln the negative charge is balanced by the positively charged amino group. Separation of the amino acids relies on the small polarity differences of the side chains: a neutral carboxyl v an amide functionality. The more repelled, negatively charged α-ketoglutaric acid has the shortest retention time.
Neutral pH & low buffer strength
At pH 6.5 there is an overall increase in polarity from the, now, fully ionised carboxyls; an increased retention for both glutamic and α-ketoglutaric acid, but not for glutamine, could be expected.
However, as the analytes carboxyls are now unprotonated and thus negatively charged, they are now repelled by the negatively charged distal sulphonates on the stationary phase. This repulsion limits the analytes ability to partition into the water layer on the stationary phase. Their retention therefore doesn’t increase as might be expected from the increase in hydrophilicity.
The only visible effect of changing the pH is that the glutamic acid is ‘moved’ to a shorter retention time due to the increased electrostatic repulsion. This resulted in all
three components being baseline separated in three minutes.
Neutral pH & medium buffer strength
Increasing the buffer strength to 30 mM effectively shields the charges on both the column stationary phase and analytes. Again the glutamine is unaffected by the change in eluent composition. None of its functional groups are affected by changing the pH, and due to the close proximity of the charges, their electrostatic interactions with the stationary phase are also unaffected when changing buffer strengths.
The effect on the two acids, especially the α-ketoglutaric acid, is however dramatic when increasing the ionic strength. It is deceptive that it has approximately the same retention time in Figures 1 and 2, but here the high hydrophilicity of this doubly negatively charged molecule is allowed to influence its retention so that it is longer retained than glutamine.
The glutamic acid elutes last, maintaining its relative elution order to α-ketoglutaric acid since it is a more polar molecule.
Discussion
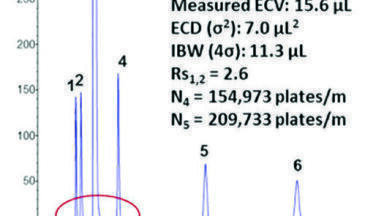
Chromatographic resolution in HILIC depends on the same factors as in reversed phase LC i.e. retention (k), column efficiency (N) and selectivity (α) (Figure 4).
As already indicated, manipulating selectivity is highly effective for resolving critical pairs in a chromatogram. Changing pH has long been used as a means of altering selectivity in reversed phase chromatography. It has even been shown that RP-HPLC at pH 2.6 and pH 10 is orthogonal enough to use for 2D chromatography of peptides4.
Changing pH in HILIC has the same effect on the ionisation state of weak acids and bases as in reversed phase chromatography. Retention will also be affected by the increased polarity of an ionised analyte but with opposite impact to reversed phase chromatography, retention will increase rather than decrease for ionised analytes. Additionally the profound effect that coulombic interactions can have in HILIC separations (a.k.a. eHILIC, ERLIC6, ion-pair normal phase7) has been illustrated here. Coulombic interactions are 10 times more powerful than hydrophilic partitioning forces (hydrogen bonding, dipole-dipole or π-π). In order to manage these interactions, a buffer is required in the mobile phase. When trying to understand elution profiles, it also worth considering that the ZIC-HILIC column is unaffected by changes in pH which can greatly simplify the interpretation of the results. This separation depends both on the ionic nature of the analytes and their coulombic interactions with the stationary phase, which differs with both eluent pH and ionic strength changes.
Resolving the complexity of the discussed separation depends on observing that the retention time for glutamine was the same under all three mobile phase conditions. Glutamine has no net charge and is thus not affected by changes in the buffer conditions. The only way to change glutamine’s retention time would be to change the acetonitrile content in the eluent.
The effect of pH changes on the carboxylic acids are two-fold, but opposing. By deprotonating the acids they become much more hydrophilic, but at low buffer strength they are also strongly repelled from the distal negative charge on the stationary phase. In order to be retained by hydrophilic partitioning the analyte needs to enter the stagnant water layer at the stationary phase surface. If electrostatic repulsion prevents this, retention will be low even though the acid is more hydrophilic. Depending on the ionic strength, deprotonating an acid could give any outcome in terms of retention; shorter, longer or unchanged. This is exemplified with the behaviour of the α-ketoglutaric acid which at low buffer strength has a slightly lower retention at pH 6.5 compared to at pH 3.0, but when the electrostatic repulsion is shielded by a high buffer strength, the retention is much higher at pH 6.5.
These experiments were undertaken using acids on a zwitterionic stationary phase exhibiting a weak anionic character. All electrostatic interactions were thus repulsive, but when separating bases electrostatic attraction would be dominant on this type of phase. By ionising a base its retention will however always increase (on the ZIC-HILIC column), both from increased hydrophilicity and through ionic attraction. Shielding the attractive forces will not reduce the retention below that which comes from polar partitioning.
Conclusion
It has been shown that by controlling the ionisation state of weak acids selectivity can be altered by changing the mobile phase ionic strength. Depending on how much salt is shielding the electrostatic repulsion between stationary phase and analyte the outcome of ionising an acid can be manipulated at will. Retention can be reduced by promoting electrosatatic repulsion or increased by shielding the repulsion and allowing hydrophilic partitioning of the more polar ionised form of the acid. By balancing these two forces, retention may also remain unchanged.
References
1. N.P. Dinh, T. Jonsson, K. Irgum, J. Chromatogr. A, 1218 (2011) 5880-91.
2. D. McCalley, "Hydrophilic Interaction Chromatography: Is it a Viable Complimentary Method to Reversed-phase for the Separation of Polar or Ionisable Compounds?" Presentation T01 at HPLC 2011, June 2011.
3. Y. Guo, et al., Chromatographia, 66, August (No. 3/4) (2007) 223-229.
4. Y. Takegawa, et al., J. Sep. Sci. 29 (2006) 2533 – 2540.
5. M. Gilar, P. Olivova,A.E. Daly, J.C. Gebler, Anal. Chem. (2005), 77, 6426–6434.
6. A. J. Alpert, Anal. Chem. 80, (2008) 62-76.
7. W. Ding, et al., Mol. Cell. Proteomics, 8, September 1, (2009) 2170-2185.




















-(1).jpg)