Bioanalytical
Published over 8 years ago. See the latest and most current information on Bioanalytical.
Small molecules still occupy a significant portion in most pharmaceutical organisation’s product list and pipeline. With the ever increasing requirement to address diseases with novel efficacious compounds, targeted quantitation of small molecule drugs is one of the most critical parts of the drug discovery and development process. Targeted quantitation enables analysis of small molecules in biological matrices to investigate efficacy and toxicity for accurate determination of the potential marketability of a drug. New methods using LC-MS/MS for the quantitation of compound mixtures in biological matrices have become more robust, accurate, and reliable, instilling higher confidence in results and furthering development of targeted drug candidates.
Inspired by chemistry, however, guided by pharmacology and the clinical sciences, small molecule (molecular weight < 1000 Da) drug research has contributed more to the progress of medicine during the past century than any other scientific factor [1,2]. However, in today’s world, owing to the increasing complexity of the diseases and hence drug targets [14,15], pharmaceutical drug discovery and development has become increasingly complex in terms of discovering and optimising new chemical entities into novel drugs with desirable efficacy and low toxicity - while ensuring compliance with regulatory requirements [3-7]. Despite the cost of development of small molecule based drugs going up constantly [4,8], they offer some advantages owing to their structural diversity. From using natural products [16,17] to new chemical entities offering novel targeted functionality, the inherent variability in small molecule based drugs still hold an important position in the world of drug discovery and development [9-12, 18-21].
Addressing regulations to ensure competency in procedures and drug safety before release to market is a critical process [13]. Such regulatory control studies require high quality and reproducible safety data for each drug tested. Regulatory agencies such as the United States Food and Drug Administration (FDA) and European Medical Agency (EMA) have regulations in place for laboratories within pharmaceutical organisations to follow in order to produce detailed analytical results supporting the development of new drugs. The typical regulatory controls include Good Manufacturing Practices (GMPs), Manufacturing and Importation Authorization (MIA) and Qualified Person (QP) declarations, among others.
Most small molecule drugs are chemically synthesised and so have a well-defined structure that can be thoroughly characterised. However, in order to test the efficacy and safety of a new drug, it must be tested and analysed in a model system. In addition to the metabolite identification (Met ID) studies, targeted quantitation is an essential piece of the drug development workflow to determine compound levels of known analytes in biological systems. Analysing drug concentration in biological tissues presents additional challenges with navigating clean extraction and obtaining an adequate yield to provide precise quantitation of compounds in a mixture. In order to resolve these challenges, robust and reliable bioanalytical methods are needed to help generate comprehensive pharmacokinetic (PK) data for drug safety analysis.
Previous targeted quantitation efforts have been performed using older generation mass spectrometry (MS) systems or ligand binding assays (LBA). While these assays are able to clearly quantify small molecules, they are unable to provide the high levels of sensitivity, selectivity, and specificity to address the complexity of the target analytes and matrices. Conversely, LBA assays are matrix and assay selective but may not have the flexibility to quantitate a wide variety of compounds or mixtures.
With advancements in both liquid chromatography (LC) and triple quadrupole MS (MS/MS), LC-MS/MS is often a preferred technology that is extensively used for targeted quantitation. LC-MS/MS offers the unique ability to systematically configure an assay specific to any molecule or mixture, as well as the ability to perform multiplexed analysis of many compounds in a single assay. LC-MS/MS also has the added benefit of using internal standards to correct for analytical variability, resulting in improved precision and accuracy of results.
In this assay, targeted quantitation was performed on a mixture of known compounds using LC-MS/MS with a Thermo Scientific TSQ Altis triple quadrupole mass spectrometer. By adding 8 small molecule compounds in pooled rat plasma to simulate the analysis of a potential drug candidate in the plasma of an animal model, LC-MS/MS can be verified for a valid and improved assay for small molecule drug candidate testing.
Rat plasma was used in combination with 8 small molecule compounds. Acetonitrile (ACN) was added to plasma in the ratio of 3:1 (v/v) to precipitate out abundant serum proteins. Although the protein precipitation clean-up process offers less efficiency in terms of yield, removal of unwanted analytes, etc. compared to some other sample preparation processes (such as solid phase extraction, etc.), this is a highly preferred option owing to its reduced cost/sample offering and increased ease-of-use. The resulting supernatant taken after processing was added to an equivalent volume of water to make the final crashed plasma stock solution. Stock solutions of each standard compound were then made at a concentration of 1 mg/mL and diluted in a pooled mix of all compounds with the crashed plasma stock to generate concentration ranges of 1 pg/mL to 25,000 pg/mL and 10 pg/mL to 100,000 pg/mL. Isotopically labelled internal standards for each compound were added to each calibration level to produce a final internal standard concentration of 0.5 ng/mL.
Chromatographic separation was performed using a Thermo Scientific Vanquish Horizon HPLC system. The column used was a Thermo Scientific Hypersil GOLD aQ C18 Polar Endcapped LC column (100 × 2.1 mm, 1.9 µm particle size). Mobile phases A and B consisted of 10 mM ammonium formate in Fisher Chemical Optima grade water (pH ~3.5), and 0.1% formic acid in Fisher Chemical Optima grade acetonitrile, respectively. The column temperature was 50ºC, with a total run time of 3.5 minutes. Chromatography gradients for analysis are summarised in Table 1.
Mass spectrometry analysis was performed on a Thermo Scientific TSQ Altis triple quadrupole mass spectrometer equipped with the Thermo Scientific OptaMax NG source housing. TSQ Altis offers several advantages over any other triple quadrupole MS with the ability to perform sensitive assays in – segmented quadrupoles, which offer better ion transmission efficiency (reduced fringe field effects), ion beam guide with neutral blocker, preventing neutrals from contaminating the quadrupoles and ensuring increased robust performance, a novel detector with increased lifetime while delivering the capability to perform robust, sensitive, fast quantitation assays.
In this study, Table 2 shows the mass spectrometer parameters and settings. Table 3 gives the selection reaction monitoring (SRM) properties used in the experimental setup.
Data analysis was performed using Thermo Scientific TraceFinder software.
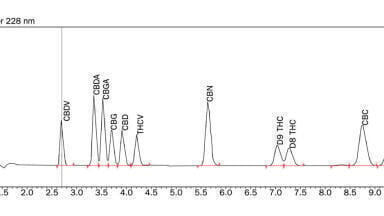
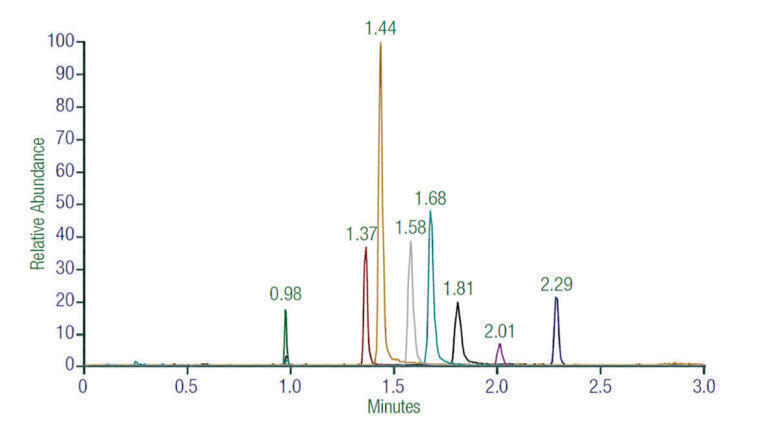
Relative abundance of each compound was determined using LC-MS/MS, providing robust and specific quantitation of the compounds isolated from the plasma matrix. Each of the eight compounds was analysed for proper detection and quantitation compared with isotopically labelled internal standards. Lower limits of quantitation (LLOQ) were obtained for each drug candidate, as shown in Table 4. In all cases, the LLOQ values were significantly lower than those observed from previous MS assay systems, suggesting excellent sensitivity across all assays. Lower coefficients of variation percentage (%CV) values for the internal standards, with four quality control points measured at increasing concentrations, also indicate improved reliability and reproducibility for tandem MS/MS. Figure 1 demonstrates high selectivity using this LC-MS/MS method.
Extended linear dynamic range is an important characteristic to studying the activity and toxicity of new compounds and mixtures. Depending on the mixture and what compounds it contains, the dynamic range can change up to, or in excess of, four orders of magnitude. This increased dynamic range might exceed the ion detector saturation limit, making the impact of ion detection significant. However, high saturation points allow for a larger dynamic range without losing low-end sensitivity. This provides the opportunity for complete pharmacokinetic (PK) profile analysis, which is essential to targeted quantitation.
In this assay, four orders of magnitude were needed in order to comprehensively cover all targets analysed, creating specific, precise, and accurate data points with no observed matrix effect. This also enables reproducibility, eliminating the need for repeated sample analysis or dilution series’ due to results being obtained outside the range. Table 5 demonstrates linearity to at least 15,000 pg/mL and high levels of reproducibility across the four quality control points taken at increasing concentrations. The method shows precision with a <8 %CV for the initial quality control point at 30 pg/mL and <3 %CV for the final quality control point at 15,000 pg/mL.
LC-MS/MS meets the increased sensitivity requirements for the analysis of small molecule pharmaceutical compounds, particularly in biological matrices where compound concentrations can be extremely low. The assay successfully achieved targeted acquisition of data, precisely detecting and measuring compound concentrations with higher sensitivity and throughput than alternative methods. The limits of quantitation for the drug candidates in plasma varied from 1-5 pg/mL, depending on the drug candidate and its components. LC-MS/MS is able to leverage increased sensitivity capabilities to achieve lower detection limits for limited sample volumes typically observed in matrix testing, due to drugs that display low tissue dispersion or have poor bioavailability.
Operating LC-MS/MS in SRM mode involves a targeted approach to identify molecules of interest and their corresponding fragments, by continuously monitoring a set of transitions for each analyte. In this manner, SRM mode generates a greater level of specificity and sensitivity of quantitation.
Combining LC and MS/MS, especially triple quadrupole MS with high resolution SRM, enables the analysis of complex mixtures with increased selectivity, where a particular molecule or group of molecules can be isolated from a complicated matrix and accurately determined to be the correct component being measured. Since compounds are separated by their mass-to-charge ratio (m/z), only those peaks appear on the chromatogram, providing enhanced selectivity of the targeted compounds. Figure 1 reflects this unmatched selectivity, showing only the clean targeted peaks of interest with no background noise and no additional unknown peaks, allowing lower detection levels as well.
Mass-to-charge ratio separation also enables LC-MS/MS to include isotopically labelled internal standards, which might pass through LC but can then be sorted by their mass difference. The use of stable isotopically labelled internal standards can help regulate variability when studying quantitation.
Analysis on a triple quadrupole instrument operated in SRM mode acts by targeted high resolution fragment ion analysis that generates a large gain in selectivity with more data points at high SRM rates. The high resolving power increases compound discrimination by separating ions of interest from interferences, thus improving the selectivity of measurements and overall quantitation function.
Matrix effect is caused by other non-targeted components in a sample affecting the analysis and quantifiable outcomes of an assay. Matrix effects and selectivity issues tend to be problematic when dealing with biological sample sources. In order to improve reproducibility and robustness of methods that are subjected to matrix effect, LC-MS/MS using a triple quadrupole instrument demonstrates significantly improved robustness of complex bioanalytical methods.
The development of a sensitive and reliable LC-MS/MS analysis method for targeted quantitation of a mixture of pharmaceutical-based compounds in biological matrices offers laboratories a novel platform for systematic, reproducible, and robust compound detection and quantitation. Applying this method to a known set of multiple drug standards suspended in rat plasma provides evidence that LC-MS/MS performed in this way can assist pharmaceutical labs with small molecule quantitation for preclinical efficacy studies.
This article is based on research by Keeley Murphy, Simon Szwandt & Claudia Martins, Thermo Fisher Scientific, San Jose, California, USA.
1. Drug Discovery: A Historical Perspective Jürgen Drews Science, 2000, 287 (5460), 1960-1964
2. Physiologically-Based Pharmacokinetics in Drug Development and Regulatory Science Malcolm Rowland, Carl Peck, Geoffrey Tucker Annual Rev Pharm. Tox., 2011, 51, 45-73
3. Optimizing drug development: Strategies to assess drug metabolism/transporter interaction potential-toward a consensus Geoffrey Tucker, Brian Houston, Shiew-Mei Huang Clin Pharmacol. & Therap., 2001, 70(2), 103-114
4. The price of innovation: new estimates of drug development costs Joseph A. DiMassi, Ronald W Hansen, Henry G Grabowski J. Health Econ., 2003, 22(2), 151-185
5. Tracking problems and possible solutions in the quantitative determination of small molecule drugs and metabolites in biological fluids using liquid chromatography–mass spectrometry Ray Bakhtiar, Tapan K. Majumdar J. Pharmacol. Tox. Meth., 2007, 55(3), 227-243
6. Deconstructing the Drug Development Process: The New Face of Innovation K I Kaitin Clin. Pharm. & Ther., 2010, 87, 356-361
7. Trends in Risks Associated With New Drug Development: Success Rates for Investigational Drugs J A. DiMassi, L. Feldman, A. Seckler, A. Wilson Clin. Pharm. & Ther., 2010, 87, 272-277
8. Estimating the cost of new drug development: Is it really $802 Million? Christopher P. Adams, Van V. Brantner Health Affairs, 2006, 25(2)
9. Targeting cancer with small molecule kinase inhibitors Jianming Zhang, Priscilla L. Yang, Nathanael S. Gray Nat. Rev. Cancer, 2009, 9, 28-39
10. Small molecule inhibitor of mitotic spindle biopolarity identified in a phenotype-based screen Thomas U. Mayer, Tarun M. Kapoor, Stephen J. Haggarty, Randall W. King, Stuart L. Schreiber, Timothy J. Mitchinson Science, 1999, 286(5441), 971-974
11. Small-molecule inhibitors of protein-protein interactions: progressing towards the dream Michelle R. Arkin, James A. Wells Nature Rev Drug Disc, 2004, 3, 301-317
12. Targeting RNA with small-molecule drugs: Therapeutic promise and chemical challenges Jose Gallego, Gabriele Varani Acc. Chem. Res., 2001, 34(10), 836-843
13. The FDA Critical Path Initiative and Its Influence on New Drug Development Janet Woodcock, Raymond Woosley Ann. Rev. Med., 2008, 59, 1-12
14. How many drug targets are there? John P. Overington Nat. Rev. Drug Disc., 2006, 5, 993-996
15.
16. Genomic sciences and the medicine of tomorrow J. Drews Nat. Biotech., 1996, 14, 1516-1518Natural products in drug discovery Alan L. Harvey Drug Disc Today, 2008, 13(19-20), 894-901
17. Drug Discovery and Natural Products: End of an Era or an Endless Frontier? Jesse W.-H. Li, John C. Vederas Science, 2009, 325(5937), 161-165
18. Raising awareness of the importance of funding for tuberculosis small-molecule research Giovanna Riccardi, Iain G. Old, Sean Ekins Drug Discovery Today, 2017, 22(3), 487-491
19. Small-molecule kinase inhibitors: an analysis of FDA-approved drugs Peng Wu, Thomas E. Nielsen, Mads H. Clausen Drug Discovery Today, 2016, 21(1), 5-10
20. Evaluating the Difficulty involved in Designing Small Molecule Drugs to Inhibit Protein-Protein Interactions, Delaram Ahmadi, David Barlow, Trends in Peptide and Protein Sciences, 2017, 1(4), 153-166
21. Development and validation of a UPLC–MS/MS method for the novel folate-targeted small molecule drug conjugate EC1456 and its metabolites in tumor homogenates from mice Satish I. Rao, Michael Pugh, Melissa Nelson, Joseph A. Reddy, Patrick J. Klein, Christopher P. Leamon J. Pharm. Biomed. Anal., 2016, 122, 148-156