LC-MS
Published over 9 years ago. See the latest and most current information on LC-MS.
Despite many recent advances in LC-MS/MS for forensic toxicology, the baseline separation of benzodiazepines is often challenging. These challenges can be met by using a high efficiency solid-core column to resolve these chromatographic interferences. Combined with a simplified solid phase extraction (SPE) protocol, accurate and precise quantification of all analytes is achieved.
High-performance liquid chromatography-tandem mass spectrometry (LC-MS/MS) is a powerful tool for quantitative analysis of drugs of abuse in the field of forensic toxicology. Many laboratories are transitioning to LC-MS/MS as they replace existing immunoassay and GC-MS based screening methods, and consolidate multiple single compound analyses into the analysis of complex compound panels. Benzodiazepines and ‘Z-drugs’ (zolpidem and zopiclone) are frequently prescribed drugs used for their sedative, anxiolytic, and hypnotic properties [1]. They work by potentiating the inhibitory neurotransmitter γ-amino butyric acid (GABA). Nationally, overdose deaths from benzodiazepines have risen 600% from under 1,600/year in 2001 to 8,000 in 2014, more than any other drug class with the exception of heroin [2].
While many laboratories have increased the use of LC-MS/MS for the analysis of benzodiazepines and other drugs of abuse in recent years, many published techniques still rely on labour- intensive liquid-liquid extraction techniques to prepare the samples .[3-5]. Some of the drawbacks of these techniques include the need to process individual samples one by one, and the use of toxic solvents. Another issue is the need to evaporate and reconstitute samples after extraction, although this can be alleviated by the use of supported liquid exchange (SLE) plates. This manuscript details an abbreviated and modified solid phase extraction (SPE) method that can rapidly extract this panel of drugs and metabolites from urine samples. It will show the benefits of having sample preparation steps, which include enzymatic hydrolysis, performed within the wells of the µElution plates. It will further show the benefits of having a water-wettable SPE sorbent that can be used without conditioning and equilibration steps. Finally, the chromatographic separation performed using a solid-core charged-surface UHPLC column will be highlighted, which enabled the baseline separation of all target analytes from internal standards with identical nominal masses. The use of a high efficiency solid core column eliminates the risk of chromatographic interference between the labelled internal standards and the native compounds.
All standards were obtained from Cerilliant (Round Rock, TX). Deuterated internal standards were used for all compounds with the exception of flurazepam. Stock solutions were prepared in methanol (MeOH). Working standards were prepared daily by diluting stock standards in 80:20 (v:v) water:MeOH. Calibrators and quality control (QC) samples were prepared in urine from working standards. High calibration standards and high QC samples were prepared in blank urine and diluted as appropriate. Each calibrator and QC sample contained all the analytes in the panel. All analytes are listed in Table 1, along with retention times and MS transitions parameters, including cone voltages and collision energies.
Sample pretreatment: 200 µL of urine was added to individual wells of an Oasis® MCX µElution Plate, along with 20 µL of internal standard (IS) solution (250 ng/mL), 200 µL of 0.5 M ammonium acetate buffer (pH 5.0) and 2 µL of β-glucuronidase (Sigma Aldrich, P. vulgate, 85k units/mL). The entire plate was incubated at 50°C for 1 hr. and then quenched with 200 µL of 4% phosphoric
acid (H3PO4).
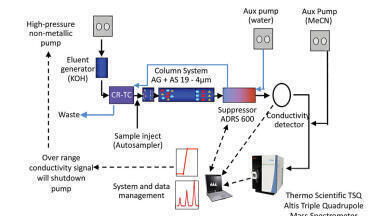
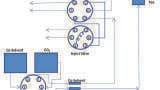
SPE Extraction: Pretreated samples were drawn into the sorbent bed by vacuum. All samples were subsequently washed with 200 µL of 0.02 N hydrochloric acid (HCl), followed by 200 µL of 20% MeOH. After washing, the plate was dried under high vacuum (~ 15 inch Hg) for 30 s. Samples were eluted with 2 x 25 µL of 60:40 (v/v) acetonitrile (ACN):MeOH containing 5% strong ammonia solution (Fisher, 28-30%). All samples were then diluted with 100 µL of sample diluent (2% ACN:1% formic acid in MilliQ water). The final concentration factor of the sample was 1.3 (200/150). A graphical workflow of the extraction procedure is shown in Figure 1.
All test compounds are listed in Table 1, and representative chromatograms are shown in Figure 1. Table 1 also lists the retention times (R.T.) and MS conditions of all compounds. The selectivity and high efficiency of the CORTECS UHPLC C18+ 1.6 µm solid-core column enables the baseline separation of all potentially interfering peaks. Two key pairs are shown in Figure 2. While clonazepam-d4 (320>274.1) (R.T. 4.08 min) generates a slight contribution to the primary lorazepam MRM (323>277), the two peaks are baseline separated. Even at the lower limit of quantitation (LLOQ) (0.5 ng/mL), the clonazepam internal standard (IS) does not interfere with lorazepam and does not affect peak quantification. Another critical pair is alprazolam-d5 and flunitrazepam. In this case, flunitrazepam (314.1>239.2) makes a contribution that can be seen in the MRM trace of alprazolam-d5 (314.1>210.1). However, the baseline separation of these peaks ensures that even at the upper limit of quantification (ULOQ) (500ng/mL) the baseline separation prevents flunitrazepam from affecting the integration and quantification of the alprazolam IS.
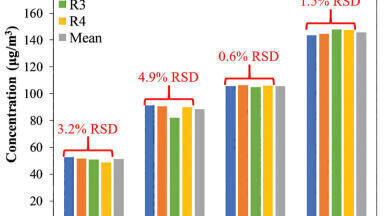
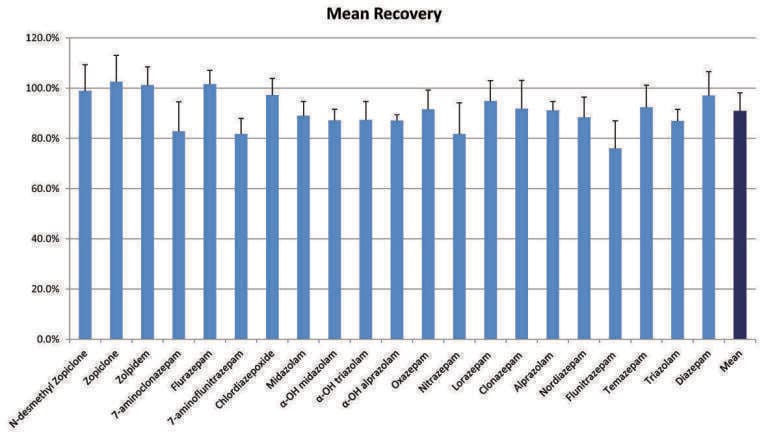
Figure 3 shows the average extraction recoveries of the entire panel of compounds from four separate experiments. All were performed at a concentration of 10 ng/mL. Recoveries ranged from 76-102% with an average of 91%, demonstrating excellent extraction efficiency. The recoveries were consistent as well, with coefficients of variation (%CVs) ranging from 5.2% to 15%, with a mean of 8.6%. The extraction method was changed from a traditional mixed cation exchange (MCX) method for basic compounds. Standard methods use wash steps, 2% aqueous formic acid followed by 100% methanol. The first wash step was modified from aqueous 2% formic acid to 0.02 N HCl to account for the low pKa’s of compounds such as clonazepam, flunitrazepam, and alprazolam and ensure ion-exchange retention on the MCX sorbent. A series of experiments performed during method development revealed that more than 20% methanol in the wash step resulted in loss of the acidic benzodiazepines, such as oxazepam, lorazepam, and temazepam. Thus, the second wash step consisted of 20% methanol, the strongest organic wash possible before breakthrough occurs. These modifications maximised reversed-phase and ion-exchange retention and enabled the highly efficient and most selective extraction of the entire panel of benzodiazepines.
Two key benefits of this method take advantage of the water-wettable SPE sorbent: the ability to directly load without conditioning and equilibration, and the ability to conduct all hydrolysis and pretreatment within the well of the SPE plate. The traditional six-step mixed-mode solid phase extraction (SPE) method was simplified into just four steps. This was accomplished by eliminating the conditioning and equilibration steps. Conducting all of the sample hydrolysis and pretreatment steps within the wells of the 96-well plate eliminates the need to transfer the sample from an incubation vessel to the SPE plate, a step that can be time consuming or error prone. After incubation within the wells of the µElution plate, the samples were simply mixed with 4% H3PO4 to quench the hydrolysis reaction and ionise the basic benzodiazepines, which were then drawn directly onto the sorbent.
Calibration curves ranged from 0.5 ng/mL through 500 ng/mL for all compounds. All compounds had LOQs of 0.5 ng/mL and upper limits of quantitation (ULOQs) of 500 ng/mL. Quality control samples were prepared at 1.5, 7.5, 75 and 300 ng/mL. A calibration summary is shown in Table 2.
Six of the curves were fitted with a 1/x weighted line, while 15 were best fit with a 1/x weighted quadratic curve. Figure 4 shows examples of compounds best fit with a line (nitrazepam, alprazolam), and a quadratic curve (diazepam, 7-aminoclonazepam). Regardless of the function used, the fits were excellent and meet the analytical needs of the method. Seventeen compounds had R2 values of 0.999 or greater, and the remaining compounds had R2 values of 0.997 or greater. Table 2 also shows that the mean % deviations for all compounds from nominal values less than 10%. Tables 3 and 4 show the results of within-batch and between-batch QC results, respectively. The within-batch results show both excellent accuracy and precision. The mean accuracies for all compounds at the four QC levels were 107.8%, 98.5%, 97.5% and 97.5%. For the highest QC values (7.5, 75, and 300 ng/mL) all individual accuracies were within 10% of target values and all %CVs were less than 10%. The between-batch results shown in Table 4 were, if anything, even better. Mean accuracies were 102.1%, 99.3%, 98.2% and 96.8% at the four QC levels. Individual co-efficients of variation (CVs) ranged from 1.1% to 9.0%. These high levels of accuracy and precision demonstrate the consistency and reliability of the Oasis MCX sorbent and extraction technique, and demonstrate that there is no compromise of result quality, even with the in-well hydrolysis and direct sorbent loading used in this method. They also show that the quadratic curves used are fit for purpose and meet the needs of the method.
A rapid and simplified solid phase extraction protocol and LC-MS/MS method for the analysis of urinary benzodiazepines and metabolites was demonstrated. Adopting this multi-analyte method provides quantitative results for a wide range of analytes in a single run. This approach significantly reduces analysis time when compared to immunoassays or single-analyte methods. Using a water-wettable SPE sorbent eliminates the common conditioning and equilibration steps associated with conventional methods without any loss in recovery or reproducibility. This property also enables the entire hydrolysis step to be conducted within the wells of the µElution plate, eliminating time consuming and error-prone transfer steps, reducing the total number of post-incubation steps from nine to five. Combining this extraction procedure with the high efficiency separation capabilities of a solid-core UPLC column, results in a rapid, efficient and exceptionally accurate analytical method.
1. Jufer-Phipps, R., B. Levine: Benzodiazepines. In: Principles of Forensic Toxicology, B. Levine (Eds). AACC Press, Washington, D.C. 237-270 (2013).
2. Karithanom, M. Number of Deaths from Prescription Drugs, National Institute of Drug Abuse, National Overdose Deaths, CDC Wonder, (2015). https://www.drugabuse.gov/related-topics/trends-statistics/overdose-death-rates
3. Laloup, M., M.d.M.R. Fernandez, G. De Boeck, M. Wood, V. Maes, N. Samyn: Validation of a Liquid Chromatography-Tandem Mass Spectrometry Method for the Simultaneous Determination of 26 Benzodiazepines and Metabolites, Zolpidem and Zopiclone, in Blood, Urine, and Hair. Journal of Analytical Toxicology 29(7), 616-626 (2005).
4. Marin, S.J., R. Coles, M. Merrell, G.A. McMillin: Quantitation of Benzodiazepines in Urine, Serum, Plasma, and Meconium by LC-MS-MS. Journal of Analytical Toxicology 32(7), 491-498 (2008).
5. Marin, S.J., M. Roberts, M. Wood, G.A. McMillin: Sensitive UPLC–MS-MS Assay for 21 Benzodiazepine Drugs and Metabolites, Zolpidem and Zopiclone in Serum or Plasma. Journal of Analytical Toxicology 36(7), 472-476 (2012).