LC-MS
Published over 7 years ago. See the latest and most current information on LC-MS.
As the technical capabilities of mass spectrometers advance - for example, greater mass accuracy and resolution - the demand for efficient analysis of increasingly complex samples via mass spectrometry (MS) has grown accordingly. Liquid chromatography (LC) and gas chromatography (GC) represent effective tools for reducing sample complexity, however co-elution of experimental components remains nearly unavoidable [1].
Though such mass spectrometer enhancements have led to greater accuracy in determining the elemental composition of sample components, their outputs lack structural information. Chemical structure data is necessary to identify sample constituents, and critical to the process of distinguishing ‘known unknowns’ - components that have been previously identified [2] - from true unknowns in MS analyses. This process, termed deformulation, typically represents a major analytical bottleneck. This is due to the significant time required to confirm the presence of all known unknowns, before moving on to isolation of any true unknowns for further characterisation.
This technical article presents a two-step deformulation approach designed to efficiently identify known unknowns by 1) utilising LC/MS/MS data to perform mass spectral searching of available libraries, and then 2) performing follow-up screening of any poorly resolved components against structural databases using predicted chemical formula and accurate mass information. This workflow utilises ACD/MS Structure ID Suite, to expedite deformulation and ensure that full elucidation activities are limited to only those components that have not been previously identified.
A metabolite identification study sample was analysed using a LC/quadrupole time-of-flight (Q-TOF)/MS. The resulting dataset was loaded into MS Structure ID Suite (v2018.1.1) for processing and analysis. A user-created MS2 spectral database was employed to perform spectral searching, followed by structure searching in local versions of the ChemSpider and PubChem structural databases, as necessary.
Within MS Structure ID Suite, the IntelliXtract algorithm (IX) was used to extract all chromatographic components. IX utilises proprietary ‘ion thread’ technology to isolate all relevant components (including differentiation of co-eluting peaks), perform peak integration, and group spectral features in order to generate a component mass spectrum. All extracted peaks were populated in the table of components (Figure 1). Spectra were annotated, and the table filled with potential confirmatory and fragment ion information where possible.
Figure 1: Table of components populated with peak data, plus pure component spectrum labelled with confirmatory and fragment ions, following sample analysis via IX.
All extracted LC/MS components were submitted for batch MS2 spectral searching simultaneously. Note that based on the variability in MS1 spectra derived from LC separations, MS2 spectra should be specified for LC/MS data, whereas MS1 data is recommended for GC/MS spectral searching. After screening a local user-created database, the table of components was further populated with the top hit for each peak found in the database, including both its structure and molecular formula if available (Figure 2).
Figure 2: The processed LC/MS dataset, with the table of components presenting the top MS spectral hit for each peak found in the database.
This database searching step can also be set to run automatically following IX analysis. Thus, the table of components would be filled with peak annotations and mass spectra as detailed following processing, plus the structure and molecular formula of each top database hit would be added.
Selecting a specific chromatographic component allows for a multifaceted evaluation of agreement between its top structural hit from spectral searching, and associated experimental data. For example, choosing the component with a retention time (RT) of 5.117 minutes in the table of components displays its corresponding pure component spectrum and MS2 data (Figure 2). The experimental MS2 is also presented alongside the database MS2 of this component’s top structure candidate in a mirrored plot for straightforward visual evaluation of hit quality. Further, MS Structure ID Suite also defines a hit quality index percentage (HQI%) to quantify the degree of candidate agreement with experimental results. For this same component at RT = 5.117 a HQI% of 74.134 was calculated, indicating a strong match. This characterisation is further supported by additional hit evaluation information from the table of components; namely, a low quantitative mass difference value (0.001 Da), and an ‘Excellent’ MS Match value (1.000).
Importantly, any component can be further examined to explore all returned database hits from spectral searching, not just the top hit as initially presented. Thus, expert users are able to manually interrogate the full complement of candidate results and replace structure assignments if necessary.
Deformulation Step 2 - Accurate Mass and Predicted Molecular Formula Screening
Molecular Formula Generation
The success of deformulation via spectral searching relies on comprehensive databases of MS1 and MS2 spectra, whether public or proprietary, and therefore components not stored previously will remain uncharacterised. One such example exists in the current dataset: a peak located at RT = 4.155 min. As this peak was not found in the local spectral database, further interrogation was required to identify it.
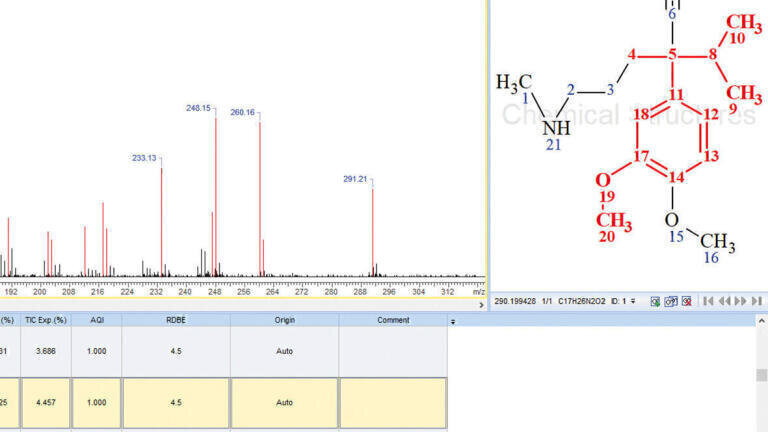
MS Structure ID Suite is well-suited for follow-up screening of such unidentified individual peaks. Examining the associated MS spectral data of this component further, the MS2 included a parent mass of 291.207 m/z. The elemental composition of this mass was then estimated, with the formula generator suggesting C17H26N2O2 as the best fit based on this component’s isotope pattern and accurate mass data.
5b. Structural Database Screening
Local ChemSpider and PubChem databases were screened for the combination of this component’s (RT = 4.155 min) parent mass (291.207 m/z) and predicted molecular formula (C17H26N2O2), while applying a tolerance of 5 ppm. The initial list of compiled structure candidates included over 35,800 hits, indicating that significant filtering was necessary to accurately identify this component. Eliminating duplicate structures trimmed the candidate list to 33,214, after which a search filter was created in MS Structure ID Suite using both a structural include and exclude list. Based on knowledge of the metabolic starting material, the correct structure for this component was expected to contain dimethoxybenzene, but not any bicyclic substructures (Figure 3). This filtering step reduced the list to a far more manageable group of 154 hits (after removal of duplicates), with a subset of candidates depicted in Figure 3, which were then examined further to discern the hit exhibiting the best agreement.
Hit Evaluation
In order to select the most suitable structure hit, all 154 candidates from the filtered list were ranked using the AutoAssignment tool within MS Structure ID Suite. This tool calculates numerical assignment scores, on a 0–1 scale, by comparing experimental MS2 spectra of the component to the candidate structure following predicted fragmentation. For the current target component, only 17 structures possessed assignment scores above 0.900. These 17 hits were further interrogated via visual examination of the complete AutoAssignment results for each candidate, to ultimately identify the structure that best matched the analytical data (Figure 4).
The newly updated deformulation workflow within MS Structure ID Suite can be efficiently and effectively used to identify multiple components from LC/MS and GC/MS datasets simultaneously, using MS2 and MS1 spectral data, respectively. The software accomplishes this task by presenting extensive, unbiased, and relevant lists of structures to identify known unknowns through spectral searching.
MS Structure ID Suite also enables streamlined characterisation of individual LC/MS and GC/MS known unknowns that are not effectively identified through spectral searching, largely due to the comparatively lesser number of known structure spectra catalogued in usable databases. The software is able to quickly search a wide range of potential structures using accurate mass and predicted molecular formulae, ensuring all known unknowns can be properly recognised before investing greater effort in elucidating true unknowns from complex samples via MS analysis.
1. Croley TR, White KD, Callahan JH, Musser SM. 2012. The chromatographic role in high resolution mass spectrometry for non-targeted analysis. J Am Soc Mass Spectr 23 (9): 1569-1578.
2. McEachran AD, Sobus JR, Williams AJ. 2016. Identifying known unknowns using the US EPA’s CompTox Chemistry Dashboard. Anal Bioanal Chem 409 (7): 1729-1735.