Identification of
biologically active substances using mass spectrometry (MS) depends on being able to eliminate spurious matches between peptides and unidentified mass spectra, according to researchers.
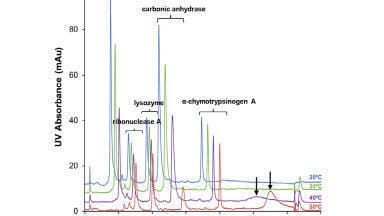
Scientists at the University of Sydney have devised an open source tool named Harvest to analyse fragmentation patterns in order to differentiate between random and correct assignments of peptides in MS-MS experiments.
This
quantitative analysis is the first time software has been developed specifically to implement and assess novel metrics for peptide matching and to explore a particular set of data's fragmentation patterns, they say.
Written in C++, the program allows fragmentation properties to be explored; however, the developers insist that it is not intended to maximise the proliferation of assigned peptides arising from a set of unknown spectra.
They also stress that the software is not for direct identification of
biologically active substances, but for the assessment of the quality of obtained data, leading to a difference between its research applications and those of similar programs already commercially available.























-(1).jpg)