Help Desk
Published over 6 years ago. See the latest and most current information on Help Desk.
The introduction of liquid chromatography mass spectrometry (LC-MS) into the analytical laboratory has transformed the ability to identify and quantify compounds at low concentrations. Initially scientists had thought that the use of this technology, which allowed for much greater specificity, would eliminate the need for any sample preparation, and the concept of dilute and shoot was readily applied to a range of samples. It was very evident that this approach has limited applicability in disciplines which require quantitive analysis as the detected levels for the same concentration of sample could vary substantially depending on the nature of the matrix components. The explanation for the variability is due to the ionisation process, which can be greatly affected by co-eluting components, or indeed the analyte itself since mass spectrometers have a limited concentration range over which they give a linear response as a function of analyte concentration. The use of sample preparation can reduce or even eliminate co-eluting species derived from the matrix which will also reduce the suppression effects caused by matrix components.
However, even when some form of sample preparation is performed, the matrix can still affect the ionisation efficiency and the performance of an assay. So called ‘matrix effects’ [1-3] are well recognised for their potential to distort the analytical data, the use of appropriate sample preparation or chromatography, however where the sample matrix varies the analyst can never be truly confident, and in this scenario the use of isotopically labelled internal standards can provide greater levels of assurance to the assay. These matrix effects arise because of the complexity of the matrix, which for a biological fluid, can contain several tens of thousands of different compounds with a very wide range (>107) of concentrations [4]. Each of the endogenous compounds can, and does, vary from sample to sample [5]. Many of these compounds will interfere with the analyte ionisation process which results in them either;
• competing for the available charge in the ion source of the mass spectrometer [6]
• enhancement of the ionisation capabilities of other compounds [7]
• reduction in solvent evaporation [8]
There are also other processes, including space charge effects, micelle formation and gas phase interactions [9] that exist and can also cause variable responses from the mass spectrometer.
The variability in matrix composition potentially means that the degree of ionisation will vary from one sample to another with possible adverse effects on the analysis of target analytes. Therefore; it is critical that the compound is resolved from any endogenous materials that produce matrix effects in order to reduce or eliminate ion suppression within the mass spectrometer source. This can be achieved either through the initial sample preparation or by the final chromatographic separation to eliminate co-elution of the matrix component and the analyte. It should be noted that in biological samples which contain tens of thousands of matrix components this will be challenging to say the least.
An interesting observation is the variability of analyte response that can be observed with the same sample and the helpdesk will look at what can cause this issue. The introduction of Incurred Sample Reanalysis (ISR) [10] as part of the validation criteria in 2009 has resulted in this issue having much greater significance and as such is a necessary component of bioanalytical method validation. ISR is intended to verify the reliability of the reported subject sample analyte concentrations and is conducted by repeating the analysis of a subset of subject samples from a given study in separate runs on different days to critically support the precision and accuracy measurements established with spiked QCs; the original and repeat analysis is conducted using the same bioanalytical method procedures.
Repeating the analysis on the same sample can potentially highlight
when there is an issue with the assay. There are a variety of reasons
that could cause the assay not to give the same result, some pertaining to the sample stability and some relating to the performance
of the assay. If the sample deteriorates over a period of time, then the assay performance should pick this up. This article will, however, focus on sample preparation issues that can affect the assay stability.
Sample Preparation
Within many bioanalytical laboratories, the typical workflow will be to perform some form of sample preparation followed by a LC-MS/MS based analysis. There are a range of different sample preparation techniques that can be employed including dilution, protein precipitation, liquid-liquid extraction, and solid phase extraction. Optimisation of each of these approaches can require some effort, making method development quite daunting. In general, the less selective the extraction technique the more economical will be the process and the quicker will be the sample preparation approach. However, the disadvantage is that there will be substantially more matrix
components that reaches the chromatographic system and ultimately this will have a detrimental effect on the performance of the system.
Two common approaches of sample preparation that are often employed are protein precipitation and solid phase extraction. Protein precipitation has been successfully applied to the analysis of a wide range of compounds within a variety of biological matrices. It relies on altering the solubility of the protein by changing the configuration of the protein using a variety of chaotropic reagents, with the most common being acetonitrile and acids such as trichloroacetic acid (TCA). Different chaotropic reagents will preferentially affect different bonding mechanisms within the protein structure. Proteins commonly cause significant issues, either due to irreversible adsorption to active surface sites on the column, co-elution or causing MS ion suppression. The removal of these matrix components increases column lifetime and also significantly reduces ion suppression effects within the detector. However, this approach does not remove all of the matrix components, and one particular classification of compounds, phospholipids, which are present in high concentrations within a biological matrix can cause high levels of ion suppression.
Figure 1 demonstrates the effect that different sample preparation techniques can have. This figure looks at the full scan spectra of a blank matrix extracted either using protein precipitation or by using solid phase extraction as a function of time with the intensity of a particular mass being highlighted by the intensity of the colour. The protein precipitation is performed using 3:1 acetonitrile to blank rat plasma (100 μL), whereas the solid phase extraction utilises a polymeric stationary phase, and washing with 30% methanol in water and eluting with 100% methanol. For the SPE method the 100 μL blank rat plasma was added to 900 μL of water prior to addition to the 1 mL 30 mg cartridge.
The chromatography was obtained on a C18 column. The mobile phases were 0.1% formic acid in water [A] and 0.1% formic acid in methanol [B]. A gradient program was used in the elution of the analytes from the column; 95% [A] and 5% [B] for 0.5 min, linear change to 5% [A] and 95% [B] over 3 min and hold for 1 min, then revert back to 95% [A] and 5% [B] and hold for 0.5 min. The flow rate was 0.6 mL/min, with the injection volume of 10 μL.
It can be seen that the protein precipitation results in a higher background level of ions, which would not be observed with many traditional assays that focus on a single parent daughter transition and do not look at a full scan spectra. In particular, there is a higher intensity of ions at longer elution times and also at the beginning of the chromatogram. Figure 2 highlights another issue with the protein precipitation approach in that it takes several aqueous blank injections before the matrix is removed. An interesting observation is that the mass spectra obtained with the first aqueous blank has higher molecular masses eluting when compared to the plasma extracted sample [11].
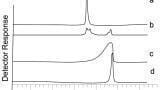
In the previous scenario the use of SPE would be beneficial to improving the robustness of the assay; however the use SPE does require a degree of dexterity to ensure that optimal performance is maintained. Figure 3 highlights one of the issues associated with SPE and one that can be quite common when dealing with multiple samples being processed simultaneously, either on a SPE manifold or using a 96 deep well (DW96) plate format. In both of these scenarios it is not uncommon to have different flow rates in different tubes/wells. There are a variety of reasons why this might exist; from poor manufacture of the SPE frits (pore structure variability etc.), to variations in the samples that are being tested, resulting in very different inter tube/well flow rates being experienced during the sample preparation step which can affect the recovery. Figure 3 shows an elution profile obtained from two samples where the flow rate has been intentionally altered to simulate this effect. It can be seen from the experiments performed that with the higher flow rate the analyte results in a greater level of breakthrough for the loading stage and that the amount of analyte that is eluted in the initial 100% methanol step is reduced, both of which have an effect on the effective recovery of the analyte.
This phenomenon is caused by the difference in time taken for the pressure driven flow compared to the time required for diffusion into the pores. Diffusion into the pore structure is required to initially capture the analyte of interest since this is where the majority of the surface area resides, thus at higher flow rates the compound simply does not get time to diffuse into the pore structure, and so analyte breakthrough is higher. During the elution part of the process the eluent is required to diffuse into the process to allow the analyte molecule to elute from the SPE media. If sufficient time is not given for this process to occur then the analyte molecule remains within the pore structure during that elution step. Robust assay development will take this effect into consideration, however the use of generic methodologies means that this is not always considered.
Chromatography
A chromatography column is designed to be used for multiple samples, and it is generally assumed for sample analysis that the chromatographic performance does not vary outside of specified performance criteria during the assay. However, it is evident that when using biological extracts that changes to the column are occuring, since the back pressure and chromatographic performance can alter throughout a batch of samples. The changes in back pressure and chromatographic performance are indicators that the surface of the column is changing and that interstitial space and/or frit porosity is being affected by matrix component build-up. Figure 4 demonstrates the effect of running a series of peptides, GSTAENAEYLR (GST), GSHQISLDNPYDQQDFFPK (GSH) and RPAGSVQNPVYHNQPLNPAPSR (RPAG) over a 6 hour period and the chromatographic deterioration that is observed. The chromatography was performed using a binary gradient from 10 - 40% of 0.025% tri-fluoroacetic acid (TFA) aq. and acetonitrile with 0.025% TFA over 10 minutes on a C18 based column. It can be seen that the peak shape deteriorates for all three components (GSH, RPAG and GST) and that there is a shift in the peak retention for one of the compounds as the stationary phase is modified.
In itself the deterioration of the stationary phase due to build-up of matrix components is detrimental, however at least in the previous example there is an obvious effect that can be seen, and so it would be possible to troubleshoot the assay with a degree of confidence in the data. A different scenario exists however when considering components that are being injected onto the chromatographic system and are not being detected, such as non-ionisable compounds, or compounds with low ionisation efficiencies under the source parameter settings. For most bioanalytical assays this is the majority of the extracted sample, with phospholipids being a good example of compounds that are not routinely detected but which can have a potential effect on the mass spectrometry. Since the elution of these components of the extracted sample are not monitored, the chromatography will not be optimised, which can result in matrix component not eluting during a single chromatographic run.
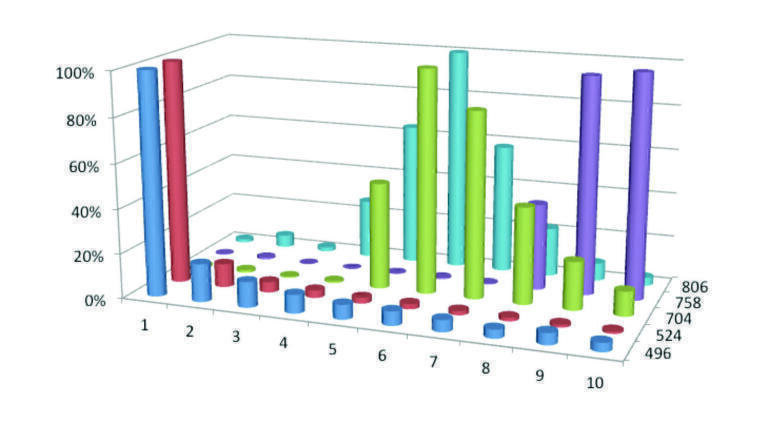
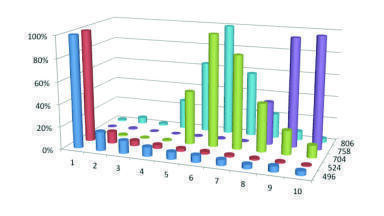
Figure 5 demonstrates this effect for a protein precipitated sample. Five phospholipid components are monitored, 1-palmitoyl-2-hydroxy-sn-glycero-3-phosphocholine, 1-stearoyl-2-hydroxy-sn-glycero-3-phosphocholine, glycerophosphocholine lipid, 1-hexadecanoyl-2-(9Z, 12Z–octadecadienoyl)-sn-glycero-3-phosphocholine and 1-(9Z, 12Z–octadecadienoyl)-2-(5Z, 8Z, 11Z, 14Z–eicosatetraenoyl)-sn-glycero-3-phosphocholines. All of these compounds have the same phosphocholine daughter group which has a characteristic mass of 184.3, with the parent masses being; 496.4, 524.4, 704.4, 758.4 and 806.4 respectively. The chromatography has been described earlier in association with the data obtained for Figure 1. It can be clearly seen that the lower molecular mass phospholipids elute in a very small number of chromatographic cycles, however the heavier molecular mass phospholipids require a substantial number of cycles to elute from the column, indeed even after 10th injection cycles some of the phospholipids are still eluting from the C18 column. The consequence of this is that the amount of suppression will vary from one injection to the next and that the amount of suppression can depend on the nature of the previous sample. Selective removal of the phospholipids will alleviate this issue, which can occur with the appropriate choice of SPE.
Conclusion
The use of sample preparation to remove matrix components is something that separation scientists need to be aware of, however it is also important to be aware of the consequences that not performing adequate sample preparation can have on the overall performance characteristics of the assay. This has greater significance within the regulated environment with the introduction of ISR, which was introduced to ensure the robustness of an assay. It has been demonstrated that the use of simple, cost effective approaches such as protein precipitation can result in greater matrix components being present in the final sample which can have a detrimental effect on the assay performance, due to the common use of mass spectrometry within a bioanalytical laboratory. Where variability is seen in a bioanalytical assay then time should be spent investigating the effects that the matrix has on the system, and then looking to address these issues through improved chromatography or the application of more selective sample preparation techniques.
In order to reduce the deleterious effects of matrix components co-eluting, it is important to be aware of the effects that the matrix components can have, and one approach is to monitor the TIC to identify when co-eluting components are coming off the column. It will also aid in determining potential types of matrix components which will allow for more selective choice of sample preparation. Phospholipids are present at high concentrations within a range of biological fluids and are renowned for causing ion suppression with a range of compounds. Monitoring these common transitions will allow the extraction process to be optimised to remove a large proportion of these components.
References
1 H. Mei, Y. Hseih, C. Nardo, et al., Rapid Commun. Mass Spectrom. 17 (2003) 97-103
2 T. Sangster, M. Spence, P. Sinclair, R. Payne, C. Smith, Rapid Comms. Mass Spectrom. 8 (2004) 1361-1364
3 B.K. Matuszewski, M.L. Constanzer, C.M. Chavez-Eng, Anal. Chem. 75 (2003) 3019-3030
4 G. Liumbruno, A. A’Alessandro, G. Grazzini, L. Zolla, J., Proteomics 73 (2010) 883-507
5 G.R. Wilkinson, Advanced Drug Delivery Reviews, 27 (1997) 129-159
6 P.J. Larger, M. Breda, D. Fraier, H. Hughes, C.A. James, J. Pharm. Biomed. Anal., 39 (2005) 206-216
7 O.A. Ismaiel, M.S. Halquist, M.Y. Elmamly, A. Shalaby, H.T. Karnes, J. Chrom. B, 875 (2) (2008) 333-343
8 R.B. Cole, A.K. Harrata, J. Amer. Soc. Mass Spectrom., 4 (7) (1993) 546-556
9 L.L. Jessome, D.A. Volmer, LCGC North America, 24 (5), 498- 510 (2006)
10 Workshop Report: Fast, D., AAPS Journal: 2009; 11: 238-241
11. F. Michopoulos, A.M. Edge, Y-T. Hui, T. Liddicoat, G. Theodoridis, I.D. Wilson, Bioanalysis 3 (24) (2011) 2747-2755




















-(1).jpg)