GC-MS
Published over 8 years ago. See the latest and most current information on GC-MS.
The importance of non-targeted analysis (NTA) methods has been steadily growing over the last several years as witnessed by an increase in publications demonstrating their utility. Several of these papers have outlined a standard protocol for the NTA portion of environmental analysis [1-3]. However most of these publications or methods have not quantifiably examined the ability to identify unknown chemicals in a sample. The purpose of the present investigation is to apply new technologies to this field of study, evaluate its ability to identify known unknowns and in so doing propose an appropriate quantifiable paradigm for identification.
There exists much precedence on identification with GC/MS from the early studies of the NIST library [4-8] and previously established United States Environmental Protection Agency (EPA) and European Union methodologies. However, many of these methods were specific to certain (classes of) compounds or did not fully embrace new technologies such as GCxGC. These procedures left gaps in quantifying the certainty of identification, which has led to several additional proposals for systematic identification [3, 9]. The Norman Association conducted the first major attempt to evaluate the methods; publishing a critical review on water analysis using high resolution mass spectrometry [3]. A total of 18 institutes in 12 countries analysed an extract of the same water sample from the River Danube and the recommendation was to produce a more comprehensive sample set for future work. Subsequently the EPA has proposed and launched a collaborative project with that goal in mind; utilising researchers who can investigate practical means to achieve confident identifications of the xenobiotic profiles within environmental samples. A round robin trial of increasing experimental complexity was designed and samples delivered to academic and industrial researchers; called the EPA’s Non-Targeted Analysis Collaborative Trial (ENTACT).
The EPA’s goals are to produce benchmark methods for analysis and reporting of non-target analysis results, standardise and facilitate future analyses, and identify areas of improvement.
The project includes 3 phases;
Phase 1: Identification of chemicals within ten mixtures which were blinded to the study participants. These blinded mixtures contained chemicals from the EPA CompTox Chemistry Dashboard [10].
Phase 2 included analysing individual standards to confirm the results from the first phase.
Phase 3 is to analyse standard references materials (SRM) that were spiked with unknown components from the mixtures.
After participants reported their identifications for Phase 1, the lists of spiked components were disclosed and the participants were allowed to re-evaluate their initial findings and submit those results as well. The overarching goals of this study are to answer the following questions:
What percentage of spiked standard mixtures are correctly identified?
Which method and processing performs best overall? Does the complexity of the mixture/matrix impact performance?
What chemical space is being covered by each method?
What can be done to expand coverage?
What unintended components (impurities, reaction, or degradation products, etc.) are in the standard mixtures?
In environmental samples, which chemicals are detected by multiple methods and are these consistencies observed in the SRM data?
The goal of this article is to report on the first phase of the trial (identification of chemicals within the ten mixtures) in our laboratory.
Our strategy for blind identification (Phase 1) was to use both electron ionisation (EI) high resolution accurate mass time-of-flight mass spectrometry (HRAM TOF-MS) and comprehensive multidimensional gas chromatography (GCxGC). To further aid in identification or confirmation of molecular ions we performed chemical ionisation (CI) experiments with the same chromatographic parameters. The data were processed according to the following steps: (1) High Resolution Deconvolution® (HRD® description below) was applied to find mathematically relevant peaks within each sample, (2) Each deconvoluted spectrum was searched against both the NIST 2017 (mainlib and replib) and a recent Cayman (v04062017) spectral library, (3) retention index (RI) values were calculated based on separate injections of a standard n-alkane mix,(4) mass accuracies of every significant m/z in each deconvolved spectrum were calculated based on formulae from the top scoring library hit. The chemical ionisation data was used solely to confirm molecular ions when needed.
The HRD algorithm treats the GCxGC-MS data as a 3-dimensional data cube populated with features or analytes which often overlap in one, two, or three dimensions simultaneously. HRD extracts these 3D features from each other and from the chemical background using a proprietary multi-dimensional deconvolution engine called Fast Accurate Robust Adaptive Deconvolution (FARAD). FARAD operates automatically without requiring preliminary knowledge of chromatographic peak shapes or an estimate of the number of coeluting components.
Experimental method parameters are described in Table 1.
Hand in hand with the experimental approach, there must be a methodology to quantify confidence in the chemical identification. Since the system used (GCxGC HRAM TOF-MS) generates a significant amount of characterising information as described above, we proposed a system which effectively assigns a confidence level for each identified compound. The proposal was loosely based on the systematic identification methodologies proposed by others, but adapted for a GCxGC HRAM TOF-MS system. To our knowledge, this is the first time this type of system has been proposed for this type of data.
Level A (Match) Every condition below must be true.
1. The forward spectral similarity score must be greater than or equal to 700 on a scale of 0-1000. Applies to EI spectra only.
2. All deconvoluted fragment m/z(s) with an abundance equal to or greater than 30% of the base m/z must have a mathematically possible formula within 5ppm based on the molecular formula of the library matched spectrum and standard valence rules.
3. A molecular ion must exist within the deconvolved spectra. It must also be within 5ppm of the expected m/z based on the matched molecular formula. If no molecular ion is present by EI, supplemental CI data can be used to provide evidence of a molecular ion.
4. The chromatographic peak’s retention index value (Kovats n-alkane scale) must be within 50 RI units of the matched library spectrum’s median experimental Semi-Standard Non-Polar RI value. If the library match does not report this value, then the RI evaluation is ignored.
5. The reviewing analyst must be confident with the peak deconvolution and identification.
Level B (Suspected Good Match)
1. Level B Suspected Match does not meet one (or more) of the Level A criteria.
2. The analyst feels there is not enough conclusive evidence to designate the peak as a Level A match, e.g. Similarity score is high but there are a number of similar compounds with identical or nearly identical spectra (isomers).
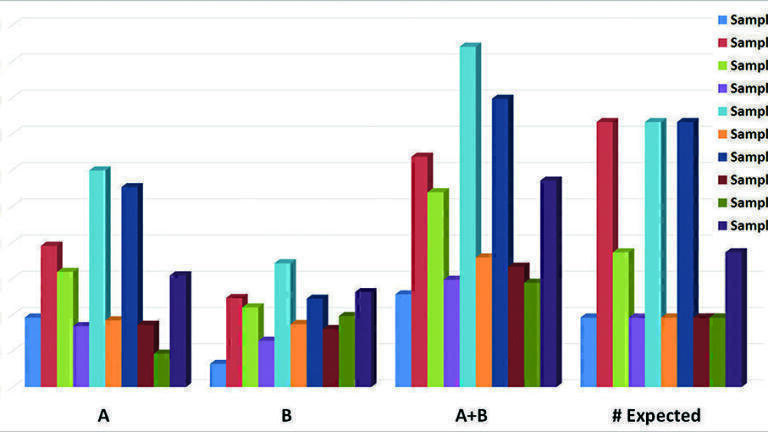
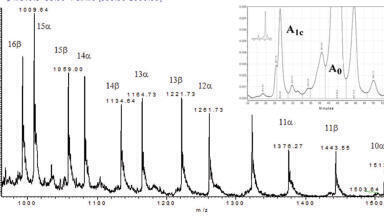
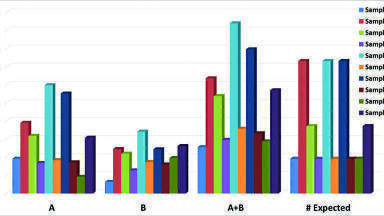
Note there were many other intense deconvoluted peaks in each and every sample but only those matching the above criteria were submitted to the EPA for review as part of the initial reporting (see Figure 1). Indeed, the advantages of the GCxGC and the highly mass accurate time-of-flight data are confirmed in Figure 1. As observed in Figure 1, the total A+B level matches was nearly always greater than the number of components expected in each sample. This would suggest the existence of unintended reaction products as described earlier. Further, the number of A level matches was also greater than the B level in nearly every sample. However, this quality measure does not indicate the accuracy of the matches. In this study, accuracy is defined by the ability to successfully identify components known to be present in the blinded mixtures (see discussion).
We divided the data review process across 5 individuals (10 samples x 2 data sets EI and CI). The results of this grading system (A’s and B’s) were submitted to the EPA as the proposed blind mixtures identification results.
After submission of the blind lists of identified components to the EPA, we received the list disclosing the standards contained in each mixture. We could then evaluate our grading system against the actual results. The matching process was carried out by matching the ‘MS-ready’ InChKey from both lists. For the most part, the matching was straight forward, a match was a match. However, there were cases where InChIKey did not match due to inconsistencies among databases so seamless, automatic matching was not possible and some manual intervention was required. Comparison of the compound list from the EPA to our largest spectral database (NIST 17) gave us our best possible results (discounting those from the Cayman library) that we could expect since our initial reporting was based solely on spectral database matching. This data is summarised in Table 2.
The maximum possible percentage of correct hits based on the NIST database varies for each sample but approximately 80% of the spiked compounds were present in NIST. There was a significant number of non-NIST compounds and those can be interpreted as laid out by others [11].
GCxGC Advantages
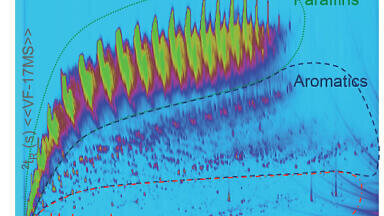
The combined GCxGC and HRAM TOF-MS approach provides multiple benefits for identification of known unknowns. A ‘known unknown’ is defined as: in the NIST MS database but unknown to be in a mixture. GCxGC is well-known to separate co-elutions which occur in simple 1D chromatography, therefore the ability to resolve more chemicals in any particular sample is increased significantly. Typically, this is quantified by the effective peak capacity (the ability to resolve chemical constituents) which is well described elsewhere [12]. Also, typically GCxGC data is displayed in what is known as a contour plot where, each dimension of chromatography is plotted (x and y axis) against total intensity as a colour (z axis). This visually appealing type of plot explicitly shows the extra dimension of resolution that GCxGC provides (Figure 2). The black dots indicate the deconvoluted peaks found by HRD algorithm. GCxGC is further known to separate analytes of interest from chemical noise and in some cases, it can also increase sensitivity over non-GCxGC experiments [13]. In Figure 2 one can observe all of these phenomena except the increase in sensitivity which is not relevant for this sample set as it was not explicitly challenged for sensitivity. One can observe the very generic nature of the experiment performed where little relevant elution occurs after 2000 seconds on the x-axis. The long hold time (30 min) was performed intentionally since this was a completely blind sample we needed to be sure to elute every component from the injection (no carry over injection to injection). Obviously the chromatography can be improved even further by using all the chromatographically available space (for more information on method optimization see Simply GCxGC [14]) and in doing so increase the accuracy of identification. This contour plot also shows column bleed peaks (banana shaped yellow-green trails), which are likely amplified due to the solvent used in the mixtures. Regardless of these challenges our results indicated a very high rate of successful identification.
In Figure 3, we examine one of many significant sections of 1st dimension GC co-elution separated by the second dimension of chromatography (y axis), each peak being separated by ≈ 100 msec. This is an interesting example since it shows how detailed the results can be for one area of first dimension co-elution. The table below the contour plot shows the match level assigned (Group), 1st dimension Retention Index (calculated), Mass Accuracy for the Molecular Ion (ppm), Formula, and Similarity score from the NIST library. Note that peaks # 893 and 902 did not have retention index information in the NIST library. Although all the other level A criteria passed it became a sub classification of the A Match. Peak # 898 was also an A level match but the deconvoluted mass spectra did not have a molecular ion, however the Chemical Ionisation spectrum did have that molecular ion evidence at the same retention time. The quality of the mass spectra generated in close second dimension GC proximity (200 msec) are shown in Figure 4 for Peaks 893 and 896. These two examples show greater than 900 similarity scores on comparison of deconvoluted to library spectra.
In Figure 5 for a particular set of ions you can see an increase number of deconvoluted peaks from 3 to 5 (increased peak capacity). Table 3 shows highlights the increased quality of the identifications obtained with comprehensive GCxGC. Undetected peaks in 1D are found in GCxGC with increased similarity scores and mass accuracies staying within the bounds of the forecasted accuracy (< 5ppm). There is just one example of many cases where this occurred the frequency of this occurrence co-elution in these samples is also the subject of a future publication.
The final topic for discussion is the overall accuracy of the identification experiment. Figure 6 shows the blind accuracy of about 84% for all samples when comparing to the NIST available compounds. Further accuracy increases to 92% upon reviewing the data after the study was unblinded. This accuracy would certainly increase after injecting the individual reference standards (future work), particularly those compounds which are missing from NIST and using them for confirmation. Or alternatively, the limitation of being constrained to those available only from NIST would be relaxed and the accuracy of identification and total percentage of identified spiked components, will only increase.
The use of a large curated database is a very significant advantage for this methodology. The latest GC/MS (EI, 70eV NIST 17 mainlib and replib) databases contain spectra for over 260,000 different chemical compounds and has been in use and validated for identification for many years [4-8]. Contrasting the GC/MS database against the ever-growing MS/MS database (for LC/MS) with spectra for greater than 15,000 compounds, truly sets the identification power of GC/MS for potential identifications over and above that for LC/MS. GCxGC HRAM TOF-MS does a superior job of identifying known unknowns at on average of 84% to the full compound list available from NIST library, as demonstrated above for these exposome standard sample mixtures.
The advantages of this approach and method are readily apparent from the percent accuracy of identification; nearly 84% correct assignments while blind is an outstanding number. Using this method practically 4 out of every 5 unknowns will be identified if they are in the NIST database. The consistency in identification across the samples and with multiple analysts is quite remarkable and indicates that sample complexity does not limit this method in its ability to identify compounds. Certainly, it is the combination of technologies that enable the methodology to be so successful. The accurate mass, high resolution data enables formula identification while the database similarity and retention index search combine to verify the known unknowns. In particular, the GCxGC technology expands the chromatographically available space significantly, improving chromatographic resolution which leads to dramatically improved peak detection compared to traditional, single dimension chromatography.
We have addressed several of the goals of the study. By identifying up to 90% of the spiked standards we believe that this method and processing performs exceedingly well and the complexity of the mixture did not impact the performance of identifications. However, we have not yet evaluated the unintended components which likely resulted in these mixtures.
Many aspects of interpretation continue to be investigated and this will be done in future work. For example, performing the same GCxGC experiments on the neat standards of each analyte (almost 4000 injections!) to take our confidence of the identification to the highest level. We are very optimistic of the accuracy of identifications once that work is complete.
The authors would like to thank Drs. Elin Ulrich and Jon Sobus (US EPA) for their efforts in initiating and coordinating this trial. Dr. Elizabeth Humston-Fulmer, Dr. David Alonso and Christina Kelly (LECO-US) provided significant assistance in data acquisition, methodology development, data review, and analysis.
References
1. Sobus, J. R., et al. (2017). “Integrating tools for non-targeted analysis research and chemical safety evaluations at the US EPA.” Journal of Exposure Science & Environmental Epidemiology.
2. Schymanski, Emma L., et al., “Identifying small molecules via high resolution mass spectrometry: communicating confidence”, Environ Sci Technol. 2014;48:2097-2098.
3. Schymanski, Emma L., et al., Non-Target Screening high high-resolution mass spectrometry: critical review using a collaborative trial on water analysis”, Anal Bioanal Chem 2015, 407:6237-6255
4. “The History of the NIST/EPA/NlH Mass Spectral Database”, Today’s Chemist at Work February 1999 Today’s Chemist at Work, 1999, 8(2), 45-46, 49-50.
5. http://www.americanlaboratory.com/913-Technical-Articles/340911-Introduction-of-NIST-17-A-Major-Update-of-Mass-Spectral-Libraries-and-Software-at-the-65th-ASMS-Conference-on-Mass-Spectrometry-and-Allied-Topics/
6. Wallace, William E., et al. “Mass spectral library quality assurance by inter-library comparison.” Journal of The American Society for Mass Spectrometry 28.4 (2017): 733-738.
7. Ausloos, P., et al. “The critical evaluation of a comprehensive mass spectral library.” Journal of the American Society for Mass Spectrometry 10.4 (1999): 287-299.
8. https://www.nist.gov/srd/nist-standard-reference-database-1a-v17
9. Sumner, Lloyd W., et al. “Proposed minimum reporting standards for chemical analysis working group (CAWG) Metabolomics Standards Iniative (MSI)” Metabolomics, 2007, 3(3), 211-221
10. EPA CompTox Chemistry Dashboard https://comptox.epa.gov/dashboard
11. McLafferty, Fred W., Turecek, Franstisek, Interpetation of Mass Spectra, 4th edition, 1993
12. Matthew S. Klee, Jack Cochran, Mark Merrick, Leonid M. Blumberg, Evaluation of conditions of comprehensive two-dimensional gas chromatography that yield a near-theoretical maximum in peak capacity gain, Journal of Chromatography A, Volume 1383, 2015, Pages 151-159
13. Lee, Andrew L., A Model of Peak Amplitude Enhancement in Orthogonal Two-Dimension Gas Chromatography, anal. Chem., 2001, 73, 1330-1335
14. Simply GCxGC. Free online tool for understanding method optimization of GCxGC www.leco.com/simply-gcxgc




















-(1).jpg)