Electrophoretic separations
Published over 13 years ago. See the latest and most current information on Electrophoretic separations.
Capillary Electrophoresis (CE) separates compounds that differ in charge to hydrodynamic size ratio and is an excellent technique for the analysis of polar compounds. The technique is particularly applicable to chiral separations. Chiral CE separation is achieved by adding a chiral selector to the so called background electrolyte. The enantiomers then form fast, reversible equilibria with the selector. In this paper a simple method development strategy for basic, acidic and neutral compounds is presented and illustrated with examples and common pitfalls. Some important good working practices (background electrolyte buffer recipes, temperature, corrected peak area, injection, polyimide coating removal from capillary ends) are highlighted, so that a good chiral separation can be developed into a robust analytical method.
Introduction
Capillary Electrophoresis has been used in the pharmaceutical industry since the first commercially available instruments appeared on the market in the late 1980s. After a decline in use during the first decade of this century, a recent revival has been observed [1], not least because of the increasing prevalence of large biological molecules as drugs. Electrophoresis has traditionally been applied mainly to proteins and nucleic acids (DNA, RNA) but CE is applicable over a wide range of analytes and anything from small inorganic ions to cell organelles and even complete cells and viruses have been analysed with CE. It is being used to complement or to replace traditional gel electrophoresis and chromatographic techniques. Typical successful applications
for small molecule are chiral as well as achiral purity determinations of drug substance and products as well as the determination of drug counter ions or small ions such as metal ions, organic acids etc. Since CE works quite well for protein analysis it has recently gained an increased interest within the biotech industry. The therapeutic proteins that are drug candidates need additional characterisation during drug discovery and development compared to traditional small molecules. Furthermore, CE methods are being used for the quality control analysis of approved biotechnological drugs. That CE has successfully gained a position in the pharmaceutical and biotech industry is demonstrated by the general chapters and monographs in pharmacopoeias e.g., the European Pharmacopoeia (Ph.Eur.) [[2], [1]] and the United States Pharmacopeial Convention (USP) [[3], [1]].
Since electrophoresis separates analytes that differ in their charge to their hydrodynamic size ratio in an applied electric field, a charge on the analyte is a prerequisite for electrophoretic mobility. Either the analyte needs to have one or more charged functional group(s) or need to form a covalent bond or a reversible complexes with either any of the background electrolyte (BGE) components or an additive (e.g., a chiral selector or a micelle). The use of additives has led to several sub techniques, such as chiral CE. It is in the field of chiral separations that CE has had its major impact in small molecule analysis. Chiral CE has the advantage of promoting highly efficient separations at a reasonable cost. In comparison, chiral gas chromatography shows effective enantioseparations but may often include a time-consuming derivatisation step. Furthermore, GC is only suitable for volatile and thermo-stabile compounds and consequently is less used today. Even though liquid chromatography (LC) and supercritical fluid chromatography (SFC) have a general high success rate for chiral separations, chiral CE is often better suited for rapid screening of optimal conditions since the enantiomeric separation is generally faster in CE and there is no need for a long equilibration time when switching to a different chiral BGE. Moreover, CE has been used to screening of selectors that later have been successfully applied as chiral stationary phases in LC [4]. With CE it is possible to screen multiple chiral selectors to a much lower cost compared to the cost for the purchase of one or more chiral columns, which reduce the expenses for the method development especially for novel compounds. Furthermore, the low solvent and solute consumption makes CE a more environmentally friendly as well as a relatively cheap technique. Finally, the high efficiency in CE means that a rather small mobility difference between enantiomers can be sufficient for their separation. So a chiral selector in CE is often more generally applicable, i.e. resolves the enantiomers of more compounds, than in LC [c.f. [5], [6]].
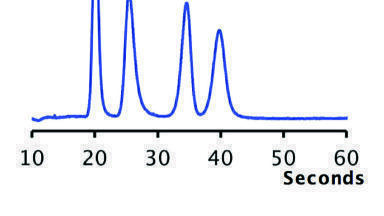
Chiral CE
Chiral CE separation is achieved by adding a chiral selector to the BGE. The enantiomers form fast, reversible equilibria with the selector. This means that part of the time an enantiomer migrates free in the BGE, and part of the time it migrates as an enantiomer-selector complex. The apparent mobilities of the enantiomers will change depending on the strength of the complex (i.e. the equilibrium constants) and the mobility of the enantiomer-selector complex. In order to separate the enantiomers, a few requirements need to be met. First of all, either the enantiomers or the selector have to be charged. Furthermore, the enantiomers of a chiral molecule need to have different affinities for the chiral selector, and/or the enantiomer-selector complexes need to differ in mobilities [[7] - [9]]. A typical example of a chiral separation is shown in Figure 1. The top trace shows an achiral CE separation of five local anaesthetic (LA) drug molecules. In the bottom trace, 10 mM dimethyl-β-cyclodextrin is added as the chiral selector to the BGE and the enantiomers of all LAs are efficiently separated [[10], [27]]. For the local anaesthetic ropivacaine, marked 3, this system was further developed into a quality control method [[11], [12]] and the method has been taken up in the Ph.Eur. and USP [[13], [14]].
Cyclodextrins (CDs) are commonly used as chiral selectors, as there is a wide variety of CDs available and they absorb little UV light at the common detection ranges. This makes CDs rather popular, although the disadvantages are that they are poorly characterised and only exist in one absolute configuration. The latter means that one cannot change the order of migration of enantiomers in a system by exchanging the chiral selector from one configuration to its
mirror image. The poor characterisation can give supplier or batch-to-batch reproducibility issues [e.g. [11], [15], [18]]. Other chiral selectors that have been used are e.g., chiral crown ethers, optically active micelles, bile salts, macrocyclic antibiotics, proteins and ion-pair selectors.
Method Development
In CE, the analyte needs to carry charge, either by itself or by complexation with e.g. a chiral selector. In chiral CE, different starting points are applied for the enantiomeric separation of basic, acidic or neutral analytes. Figure 2 sketches our experience for a typical starting situation. The strategy to select a starting point for a basic analyte is usually relatively simple. At a low pH, at least two pH units below a basic analyte’s pKa, it is mainly (≥ 99%) charged and at a low pH (i.e. below pH 4) also the electro-osmotic flow (EOF) is low. This means, that either a charged or an uncharged chiral selectors can be used. The advantage of using an uncharged selector is that it does not contribute to the ionic strength of the electrolyte and therefore does not increase the current and Joule heating. An uncharged selector migrates with the electro-osmotic flow and the separation window for the enantiomers is between the migration of the unaffected analyte, i.e. the migration velocity
of the enantiomeric analytes with no selector present, and the EOF (Figure 3). For basic analytes with a BGE at a low pH and slow EOF, the separation window is rather large. The advantage of a negatively charged selector is that it migrates slower than or in reverse direction of the EOF, which enlarges the separation window further. The disadvantage of a charged selector is that it can contribute considerately to the ionic strength of the system, resulting in (often too) high currents.
For acidic compounds, the strategy to obtain enantiomeric separation requires usually more attention. At a high pH, over 2 pH units over the pKa, the analyte is charged but a high pH (i.e. pH ≥ 8) also gives a fast EOF. The high EOF decreases the time it takes for the analyte to reach the detector. The separation window for the analyte becomes rather small in case of an uncharged selector migrating with the EOF (Figure 4). One possibility to improve the separation window is to use a charged selector. Although there are alternative solutions to improve the separation window when there is no appropriate charged selector for the enantiomers at hand. An increase in the separation window can be obtained in the case of an uncharged chiral selector e.g., by using a coated capillary or by influencing the migration of the free enantiomer, such as with CD MEKC (explained in Figure 4) or with a dual CD system (explained in [[16], [17]]). An interaction or partition to an achiral micelle or with a charged CD, that not necessarily exhibit stereoselective interaction with the analyte, will promote a migration of the enantiomers delayed from the EOF. This delay increases the separation window.
For the separation of neutral analytes, the selector must be charged or the charge has to be induced on the enantiomers with CD-MEKC or a dual CD system similar as mentioned above for acidic enantiomers.
Once a selector that gives enantioselective interaction for the chiral analytes is found, the separation can be optimised. An important factor affecting the separation is the concentration of the chiral selector, as the concentration will determine the equilibria between the selector and the enantiomers. Often pH, temperature, buffer composition or the magnitude of the EOF can also affect the enantiomeric separation so these parameters should be investigated as well, preferably in a multifactorial design. In the literature there are many examples both for method optimisation as well as robustness testing applying chemometrics [e.g. [12], [19] - [25]]. An additional factor that needs attention during robustness testing is the purity of the chiral selector. Often batch-to-batch variability results in a change in chiral resolution. The robustness test should include testing of selectors from different suppliers and batches and result in a system suitability test (SST) with appropriate requirements, such as resolution.
Pitfalls and Examples
It is key to find the right chiral selector. The most important point here is not to be prejudiced. Because of the high efficiency in CE, you need very little difference in selector interaction between the enantiomers in order
to obtain chiral resolution. For example, although it is often stated that you need an aromatic group to resolve enantiomers with cyclodextrin as chiral selectors, it was demonstrated that some non-aromatic dipeptides could be separated with several different uncharged CDs [[18][13],[26]].
A commonly made error is that too few selectors are tested during the initial screening phase. As demonstrated in Figure 5, it is very hard to predict separation. The 16 different forms of a tetrapeptide, sorted as eight enantiomeric pairs, resolved very differently in the same system. A screen of different CDs for separation of these tetrapeptide enantiomers gave different results for each CD (Table 1).
However, sometimes the highest resolution from an initial test will not necessarily lead to the optimum separation. For the enantiomeric separation of adrenaline, which can be present in local anaesthetic (LA) solutions, a screen of suitable selectors was performed. The aim was to obtain enantiomeric separation of adrenaline, preferably undisturbed by the presence of any of the LAs that could be present in the injection solution (Figure 6), in order to investigate racemisation of adrenaline during shelf life. The highest resolution for adrenaline was obtained with HDAS-β-CD, but the best separations were with DM-β-CD. In the latter system, rac-adrenaline was well separated and migrated before the LAs. The LAs are present in 1000x higher concentrations than L-adrenaline in injection solutions, and with the migration order in the system using DM-β-CD the disturbance on adrenaline of the LAs was least. The example also demonstrates the importance of working with real samples in method development and validation.
Good Working Practice
To move on from a good separation to a robust and sensitive method, we need to adopt good working practices on top of fundamental knowledge and understanding of the technique and its modes of operation. While all aspects of CE good working practice cannot be discussed here, there are some important points worth highlighting.
Electrolyte System
For all CE methods, chiral or achiral, it is paramount to describe the electrolyte system unambiguously. This is best done by describing the exact concentrations for the electrolyte components. For example, “make a solution containing 100 mM phosphoric acid and 88 mM triethanolamine. Check the pH, which should be pH 3.0 ± 0.1” is to be preferred over “100 mM phosphoric acid adjusted to pH 3.0 with triethanolamine”. As long as the electrolyte is buffering (which it should) one drop more or less of the triethanolamine does not change the pH very much, but it does affect the ionic strength of the system. Better reproducibility is obtained when the ionic strength is controlled. Consequently, one should use appropriate volumetric glassware.
Although the above mentioned phosphate triethanolamine electrolyte still has some buffering capacity at pH 3.0, this sometimes is not sufficient [[18]]. When the analytes have pKa values close to the pH of the buffer and when the buffer capacity is low, problems can arise. Injection of a dipeptide in phosphate buffer pH 3.0 gave enantiomeric resolution, which disappeared with repeated injections. This problem was easily solved by exchanging phosphoric acid (pKa 2.15) to malonic acid (pKa 2.85) as buffering component. The change in pH due to the electrolysis of water in phosphoric acid - NaOH buffers after applying voltage is illustrated in Table 2. The best buffering capacity is obtained at as high buffer concentration as possible and with a pH within one pH unit from the pKa [[28], [29]] of the buffer protolyte. The buffering capacity steeply reduces the more the pH deviates from the pKa value of the weak acid in
the buffer.
Temperature
The temperature control of a CE capillary is important. Temperature influences parameters such as the viscosity of the BGE and pH and pKa values of buffers and analytes which influences the chiral as well as achiral equilibria in the BGE. For good repeatability and reproducibility, temperature therefore needs to be carefully controlled. In chiral systems, temperature may also influence the degree of complexation of the enantiomers with the chiral selector. Decreasing the temperature can either increase or decrease the resolution [[30]].
Corrected Peak Areas
Altria [[33]] demonstrated by the separation of a racemic mixture of picumeterol that it is important to correct the peak areas of the separate enantiomers for their migration velocity difference through the detection window. Early migrating analytes move faster through the detection window than later migrating analytes and thus show up as smaller peaks. Therefore the peak areas are corrected by either dividing by the migration time or by the migration velocity (Table 3).
Injection
For good precision in chiral CE, the same attention to detail is needed as for other CE techniques [[31]]. That is, a constant temperature on the samples and the BGE. As short injection time as possible above 3 s should be used. This is because in some CE instruments the variability in hydrodynamic injection volume is reduced by stringent control of the injection time x injection pressure. For this mechanism to function properly, at least 3 s injection time is needed. The vials should be levelled to prevent siphoning. Excess of sample sticking on the outside of the capillary after injection can be removed by dipping the capillary inlet after injection into a vial filled with water or BGE. The precision can be further increased by injection of a BGE or water plug after the injection of the sample [[31]]. Around two millimetres of the polyimide coating should be removed from the capillary inlet whenever possible, and of course straight capillary ends [[32]]. An enantiomeric purity determination often requires detection as low as 0.05 % of the enantiomeric impurity. In order to obtain these levels of sensitivity, it is often needed to use sample stacking. The simplest approach is to dissolve/dilute the sample in a less conducting solvent compared to the BGE. When the voltage is applied, the low-conductivity sample zone will have a higher electric field strength than the BGE. Consequently the analytes migrate faster in the injection solution zone until they reach the boundary of the BGE zone. In the more conductive BGE the field strength is lower and the analytes stack, i.e. slow down and concentrate, which reduces the initial band width. However, many other sample stacking techniques are also available, see e.g. [[34]].
Concluding Remarks
Capillary electrophoresis has developed into a highly efficient separation technique and is currently applied in several different areas of analysis such as chiral separations. Further implementation of CE and chiral CE in industry depends on sharing and training of good CE working practices and access to the industrial expert network, as well as on continuous development of instrumentation [[1]].
Capillary electrophoresis made a significant contribution to the Human Genome Project and is emerging as an import analytical tool in the area of metabolomics and other
‘-omics’. In these areas the objective is to simultaneously analyse a large number of hydrophilic compounds such as amino acids, peptides and other analytes that might be present as enantiomers. Chiral CE shows high separation efficiency and is suitable for hydrophilic analytes and might thus be the preferred technique. A key factor for success is CE-MS. Unfortunately, the combination of CE and MS has not yet been fully established to be used routinely in the laboratory. When hyphenated CE-MS/MS becomes an analytical tool as ordinary as LC MS/MS is today, it will certainly make CE an even more attractive separation technique in several areas. In chiral CE, it would be preferable to apply the partial filling technique to avoid the selector to reach the MS instrument [[35]] and thus minimise time-consuming cleaning of the MS interface.
Automation and miniaturisation of analytical systems have been and still are general trends in analytical chemistry. The use of a multi-capillary CE system has proven to considerably improve the sample throughput [e.g. [36][37]]. Interesting studies applying electrophoretic microfluidic devices for fast chiral separations have been published [[38], [39]] and more studies will be published in the future.
References
[1] C.E. Sänger – van de Griend, R.E. Majors, LC-GC North America 30 (2012) 954
[2] General Chapter 2.2.47 Capillary Electrophoresis, European Pharmacopoeia
[3] General Information <1053> Capillary Electrophoresis, United States Pharmacopeia
[4] V. Piette, M. Lammerhofer, W. Lindner, J. Crommen, J Chromatogr A 987 (2003) 421
[5] Y. Carlsson , M Hedeland, U. Bondesson , C. Pettersson, Chromatogr. A 922 (2001) 303
[6] C. Petterson, C. Gioeli, Chirality 5 (1993) 241
[7] S.A.C. Wren, R.C. Rowe, J. Chromatogr. 603 (1992) 235
[8] S.A.C. Wren, J. Chromatogr. 636 (1993) 57
[9] B. Chankvetadze, Capillary Electrophoresis in chiral analysis, John Wiley & Sons, 1997, ISBN 0 471 97415 3
[10] C.E. Sänger – van de Griend, K. Gröningsson, D. Westerlund, Chromatographia 42 (1996) 263
[11] C.E. Sänger – van de Griend, K. Gröningsson, J. Pharm. Biomed. Anal. 14 (1996) 295
[12] C.E. Sänger – van de Griend, H. Wahlström, K. Gröningsson, M. Widahl-Näsman, J Pharm. Biomed. Anal. 15 (1997) 1051
[13] 2335 Ropivacaine Hydrochloride Monohydrate, European Pharmacopoeia
[14] Monograph Ropivacaine Hydrochloride Monohydrate, United States Pharmacopeia
[15] A. Amini, T. Rundlöf, M.B. Grön-Rydberg, T. Arvidsson, J. Sep. Sci. 27 (2004) 1102
[16] G. Gübitz, M.G. Schmid, Cyclodextrin mediated chiral separations, in Chiral Separations by Capillary Electrophoresis, Ed. A. Van Eeckhaut, Y. Michotte, CRC Press, 2009, ISBN 978-1-4200-6933-4
[17] A.C. Servais, J. Crommen, M. Fillet, Factors influencing cyclodextrin mediated chiral spearations, in Chiral Separations by Capillary Electrophoresis, Ed. A. Van Eeckhaut, Y. Michotte, CRC Press, 2009, ISBN 978-1-4200-6933-4
[18] C.E. Sänger – van de Griend, Electrophoresis 20 (1999) 3417
[19] P.K. Owens, A.F. Fell, M.W. Coleman, and J.C. Berridge, Chirality 9 (1997) 184-190.
[20] S. Boonkerd, M.R. Detaevernier, Y. Vander Heyden, J. Vindevogel, and Y. Michotte, J. Chromatogr. 736 (1996) 281
[21] S. Fanali, S. Furlanetto, Z. Aturki, and S. Pinzauti, Chromatographia 48 (1998) 395
[22] M.M. Rogan, K.D. Altria, and D.M. Goodall, Chromatographia 38 (1994) 723
[23] M.I. Jimidar, T. Vennekens, W. Van Ael, D. Redlich, M. De Smet, Electrophoresis 25 (2004) 2876
[24] V. Harang, M. Tysk, D. Westerlund, R. Isaksson, G. Johansson, Electrophoresis 23 (2002) 2306
[25] X. Deng, B. De Cock, R. Vervoort, D. Pamperin, E. Adams, A. Van Schepdael Chirality 24 (2012) 276
[26] C.E. Sänger – van de Griend, Electrophoresis 21 (2000) 2397
[27] Enantiomeric separations by capillary electrophoresis in pharmaceutical analysis, by C.E. Sänger – van de Griend, Uppsala University Library, 1999, ISBN 91-554-4561-6
[28] R.J. Beynon, J.S. Easterby, Buffer solutions, Oxford University Press, 1996, ISBN 0-19-963442-4
[29] B.M. Thomson, M. A. Kessick, J Chem Educ 58 (1981) 743
[30] A Westall, T Malmström, P Petersson, Electrophoresis 27 (2006) 859
[31] Injection precision and sensitivity, CE Solutions issue 5, http://www.sepscience.com/Techniques/CE
[32] The CE capillary, CE Solutions issue 3, http://www.sepscience.com/Techniques/CE
[33] K.D. Altria, Chromatographia 35 (1993) 177
[34] M.C. Breadmore, A.I. Shallan, H.R. Rabanes, D. Gstoettenmayr, A. Syazwani Abdul Keyon, A. Gaspar, M. Dawod, J.P. Quirino, Electrophoresis 34 (2013) 29
[35] H. Lodén, Y. Hedeland, M. Hedeland, U. Bondesson, C. Pettersson, J. Chromatogr. 986 (2003) 143
[36] S. Behr, M. Mätzig, A. Levin, H. Eickhoff, C. Heller, Electrophoresis 20 (1999) 1492
[37] M. Herrero, C. Simó, V. García-Cañas, S. Fanali, A. Cifuentes, Electrophoresis 31 (2010) 2106
[38] M. Ludwig, F. Kohler, D. Belder, ”High-speed chiral separations on a microchip with UV detection”. Electrophoresis 24 (2003) 3233
[39] S. Nagl, P. Schulze, M. Ludwig, D. Belder “Progress in microchip enantioseparations” Electrophoresis 30 (2009) 2765
[40] C.E. Sänger – van de Griend, K. Gröningsson, T. Arvidsson, J. Chromatogr. A 782 (1997) 271
[41] C.E. Sänger – van de Griend, A.G. Ek, M.E. Widahl-Näsman, E.K.M. Andersson, J. Pharm. Biomed. Anal. 41 (2006) 77