Electrophoretic separations
Published over 5 years ago. See the latest and most current information on Electrophoretic separations.
The analytical approaches mainly used in metabolomics for addressing biological questions are based on liquid chromatography-mass spectrometry (LC-MS) and nuclear magnetic resonance (NMR) spectroscopy. However, the value of metabolomics, which in essence is obtaining insight into a well-defined biological problem, may be completely overlooked when only these analytical technologies are considered. Notably, for biological questions intrinsically dealing with low sample amounts, but also for the study of ‘difficult’ compound classes, such as low-abundance highly polar ionogenic metabolites. This work aims to highlight the possibilities of capillary electrophoresis-mass spectrometry (CE-MS) for metabolomics by paying attention to some key fundamental and technological aspects to address perceived misconceptions about this analytical technique. With recent examples, we show the utility of CE-MS for special applications and thereby the value of this approach for metabolomics.
Compared to other analytical techniques, the use of CE-MS is underrepresented in metabolomics [1], presumably as this analytical technique is perceived as less reproducible, in particular for migration time, by the separation science community [2]. In metabolomics studies, migration-time reproducibility is of pivotal importance as it ensures a reliable comparison of metabolic profiles, including scrutinising samples for subtle changes in patterns. Moreover, it supports the identification of unknown metabolites and is considered complementary to high-resolution MS/MS.
In CE-MS-based metabolomics studies, variability in migration time mainly arises from fluctuations in the electro-osmotic flow (EOF) caused by sample matrix-induced capillary surface interactions. Recently, González-Ruiz et al. addressed this challenge by developing software, designated as ROMANCE, which converts the migration-time scale into an effective electrophoretic mobility scale [3]. The approach is based on utilising the fundamental separation principle of CE (in this case specifically referring to capillary zone electrophoresis), i.e. the effective electrophoretic mobility of the solute, which in essence depends on the charge and size of each compound (assuming other factors to be constant such as viscosity of the separation buffer). ROMANCE allowed effective correction of migration-time shifts caused by the EOF and as a result, improved the repeatability of the CE-MS analyses. By using this approach based on effective electrophoretic mobility, Drouin et al. constructed a library for 458 endogenous metabolites in order to facilitate metabolite identification by CE-MS [4].
To assess the true power of using effective electrophoretic mobility in CE-MS-based metabolomics studies, Drouin et al. recently set-up a Metabo-ring trial to which 13 independent laboratories from 11 countries contributed [5]. All laboratories used the same batch of samples, comprising representative metabolite mixtures, human plasma and urine spiked with the same representative metabolites. Each participating lab prepared and employed the same background electrolyte (BGE) based on a protocol. All other parameters, i.e. capillary, interface, injection volume, voltage ramp, temperature, type of instrument used, capillary conditioning and rinsing procedures, etc., were left entirely to the participating labs’ discretion. The critical parameters examined by this Metabo-ring were the reproducibility of relative migration time and effective electrophoretic mobility across the laboratories, which was determined for a set of cationic metabolites in each sample. Despite the huge heterogeneity in experimental conditions and platforms across the 13 independent labs, conversion of migration times into effective electrophoretic mobility reduced variability from 10.9% on relative migration time to 3.1% in effective electrophoretic mobility scale using the same BGE composition. Although this work primarily focused on cationic metabolic profiling, it is anticipated that the same strategy could also be applied to anionic metabolic profiling. Overall, this study, which could be regarded as unique given its design, clearly exemplified CE-MS’s actual reproducibility in metabolomics, i.e., effective electrophoretic mobility can be used as a robust parameter in metabolomics.
It should be noted that CE-MS has been used for more than two decades to investigate urinary peptides as biomarkers for the diagnosis and prognosis of (complex) diseases [6]. Until now, the CE-MS peptidomics approach was employed for the comparable analysis of more than 70000 urine samples and has previously been qualified for prognosis of progression and outcome in large-scale prospective and longitudinal clinical studies [7, 8]. In Germany, the CE-MS peptidomics approach has been registered now as an in vitro diagnostics for a number of clinical applications [9].
The coupling of CE to MS is often perceived as a technically challenging endeavor, notably when compared to LC-MS or GC-MS [10]. The lack of standard operating procedures and data workflows that are fit for purpose may also have hindered the widespread use of CE-MS in metabolomics despite new advances in sample throughput and quality control [11, 12]. For cationic metabolic profiling, well-established CE-MS protocols have been developed over the past decades and employed to analyse large sample cohorts, such as the Tsuruoka Metabolomics Cohort Study, comprised of more than 8000 human plasma samples [13]. The CE-MS approach utilised for this metabolomics study was provided by Human Metabolome Technologies (HMT), a Japan-based biotechnology company founded by Soga and coworkers at Keio University. They developed the first CE-MS methods for metabolomics [14]. Today, the CE-MS approach of HMT for cationic metabolic profiling can be used in a robust way and currently employed by various research groups. However, the development of a robust CE-MS approach for anionic metabolic profiling is still an ongoing development. For example, Yamamoto et al. recently showed that commonly employed ammonium-based BGEs with a pH above 9.0 could contribute to incidental capillary fractures via irreversible aminolysis of the outer polyimide coating [15]. The study revealed that polyimide
aminolysis could be simply prevented by employing weakly ammonium-based BGEs with a pH below 9.0.The previous example, and some other recent work, clearly show the effort and willingness of the CE-MS community to highlight relevant technological and practical aspects for metabolomics studies in protocol papers [16-20]. An important recent trend in this context is sharing key experimental procedures and best practices via peer-reviewed video articles [21-24], and it is anticipated that such work will encourage researchers to actively consider CE-MS for metabolomics studies. Worthwhile to mention in this context is that the recent CE-MS Metabo-ring trial clearly revealed that this approach can be used in a rather straightforward way even by groups without having (any) experience with metabolomics research [5].
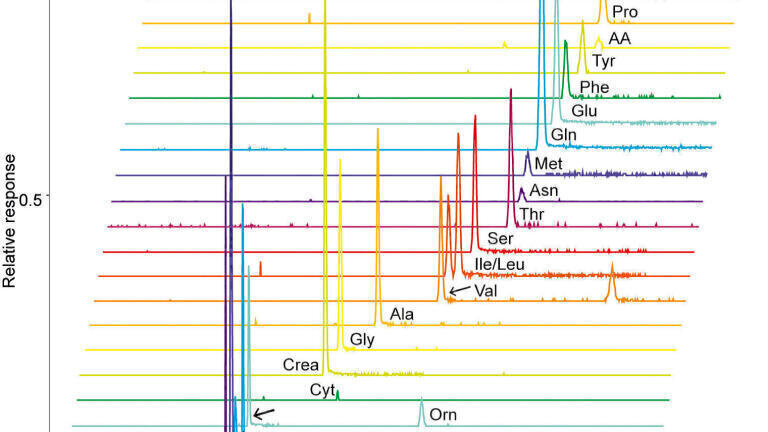
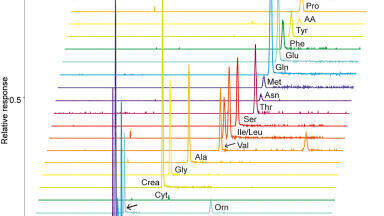
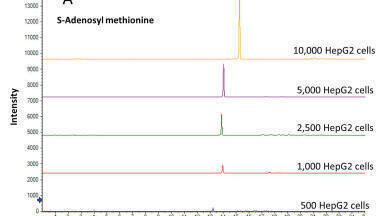
The prerequisite of low sample volumes for CE-MS analysis makes it an attractive tool for the analysis of volume-limited or scarcely available samples. However, due to the limited loading capacity of CE and concentration-sensitive detection of ESI-MS, the technique is often perceived as non-suitable for trace metabolomics. Nevertheless, the loading capacity of CE has been addressed effectively by the use of in-capillary sample preconcentration techniques. Recently, Wells et al. applied preconcentration based on electrokinetic supercharging for neurotransmitter analysis in volume-limited tissue samples from rat brain tissue whole Drosophila [25], thereby reaching detection limits as low as 10 picomolar. In another study, van Mever et al. optimised the use of dynamic pH junction stacking for rat brian microdialysis samples [26], thereby showing the compatibility of the stacking procedure with the high-salt matrix of the microdialysate (Figure 1). Detection limits were in the low nanomolar range for amino acid neurotransmitters, showing its potential for trace-sensitive brain metabolomics studies.
Additionally, it should be noted that in both previous discussed works, a conventional sheath-liquid ESI CE-MS interface was used. When using a sheathless CE-MS interface, a 10-fold improvement of detection sensitivity could be achieved. Thus, CE-MS could definitely yield comparable detection limits as compared to conventional LC-MS methods, with the main difference being that for CE only a volume of about 30-300 nL is injected, while 5-10 µL in the sample vial is sufficient for injection, whereas typically 300-10000 nL is injected in a conventional LC-MS method. This makes using CE-MS beneficial when the sample amount is very limited, such as for example is the case for single cell analysis. Recently, the potential of CE-MS for single cell analysis has been reported repeatedly [27] and Lombard-Banek et al. reported a CE-MS method that allowed in vivo single-cell proteomics and metabolomics in the same single cell in chordate embryos using X. laevis [28]. With a custom-build CE-MS system, quantitative proteo-metabolomic differences were observed between cells at the cleavage stage. This work shows the potential of CE-MS for trace-sensitive metabolomics, including the ability to study cell heterogeneity in future metabolomics studies.
High-throughput analysis of dozens, hundreds or even thousands of biological samples is gaining importance for metabolomics studies. Especially there is a requirement for fast and robust metabolomics workflows for volume-restricted samples. A notable improvement in CE-MS analysis strategies is the multi-segment injection (MSI) approach [29], developed in 2013 by the research group of Britz-Mckibbin. MSI allows for serial injections of seven or more samples within a single capillary, thereby significantly improving the sample throughput. Furthermore, when including a quality control sample, stringent quality control and batch correction can be performed during the same run. In the last few years, MSI-CE-MS has been an efficient analysis tool for metabolomics studies in various sample types and CE-MS methodologies [30-32].
Recently, MSI-CE-MS potential for large-scale metabolomics was shown in a study including more than a thousand serum samples [20]. In this study, metabolic profiles in serum samples from pregnant woman all over Canada were analysed for 7 months using standardised methodology and data treatment. The results showed acceptable intermediate precision for a range of metabolites. Overall, this work clearly demonstrated the value of MSI-CE-MS for executing in a robust way large-scale high throughput metabolomics studies, including successful correction for long-term signal drift and inter-batch variations.
To date, the vast majority of CE-MS-based metabolomics reports have focused on the analysis of polar ionogenic metabolites using an aqueous buffer system that may include small amount of organic solvent modifier (5-10% v/v) using CZE as the main separation mode. CZE-MS is ideal for the profiling of diverse classes of highly polar metabolites (including, organic acids, nucleotides, sugar phosphates) and their intact conjugates (e.g., glycine, sulfate or glucuronide) that are poorly retained on reversed-phase LC or undergo excessive band broadening in hydrophilic interaction LC analyses. Next to CZE, there are other CE separation modes showing potential for metabolomics studies, such as non-aqueous CE (NACE) and micellar electrokinetic chromatography (MEKC). NACE, in which background electrolytes (BGEs) are composed of organic solvents containing volatile salts such as ammonium acetate in a small portion of water, has interesting features for the analysis of apolar and charged compounds. Moreover, the use of high organic solvent-based BGEs may further improve the electrospray ionisation efficiency. Recently, Azab et al. developed a NACE-MS method to profile more than 20 non-esterified fatty acids in human plasma and serum [33]. The NACE-MS approach in conjunction with MSI allowed rapid yet comprehensive profiling of fatty acids in volume-restricted samples.
MEKC, first introduced by Terabe and coworkers [34], can be used for the separation of neutral and charged compounds. In MEKC, ionic micelles or surfactants, often SDS, are used as pseudo-stationary phase and the separation is based on the differential partition of neutral and charged compounds between the micellar phase and the aqueous BGE. Given the nonvolatile nature of SDS, the on-line coupling of MEKC to MS is challenging since the introduction of nonvolatile surfactants into the MS may decrease ESI efficiency (ion suppression) and contaminate the ion source. To tackle this issue of incompatibility, Moreno-González et al. developed a selective and sensitive MEKC-MS method employing ammonium perfluorooctanoate (APFO) as volatile surfactant for the analysis of amino acids in human urine [35]. This method was further optimised by Prior et al. and used for the enantioselective analysis of amino acids in cerebrospinal fluid [36]. It is anticipated that NACE and MEKC will further expand the role of CE-MS in metabolomics in the coming years.
Marlien van Mever and Rawi Ramautar would like to acknowledge the financial support of the Vidi grant scheme of the Netherlands Organization for Scientific Research (NWO Vidi 723.016.003).
The authors have no other relevant affiliations or financial involvement with any organisation or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
1. Miggiels, P., Wouters, B., van Westen, G. J. P., Dubbelman, A.-C., Hankemeier, T., TrAC Trends in Analytical Chemistry 2019, 120, 115323.
2. Buscher, J. M., Czernik, D., Ewald, J. C., Sauer, U., Zamboni, N., Anal Chem 2009, 81, 2135-2143.
3. Gonzalez-Ruiz, V., Gagnebin, Y., Drouin, N., Codesido, S., Rudaz, S., Schappler, J., Electrophoresis 2018, 39, 1222-1232.
4. Drouin, N., Pezzatti, J., Gagnebin, Y., Gonzalez-Ruiz, V., Schappler, J., Rudaz, S., Anal Chim Acta 2018, 1032, 178-187.
5. Drouin, N., van Mever, M., Zhang, W., Tobolkina, E., Ferre, S., Servais, A. C., Gou, M. J., Nyssen, L., Fillet, M., Lageveen-Kammeijer, G. S. M., Nouta, J., Chetwynd, A. J., Lynch, I., Thorn, J. A., Meixner, J., Lossner, C., Taverna, M., Liu, S., Tran, N. T., Francois, Y., Lechner, A., Nehme, R., Al Hamoui Dit Banni, G., Nasreddine, R., Colas, C., Lindner, H. H., Faserl, K., Neususs, C., Nelke, M., Lammerer, S., Perrin, C., Bich-Muracciole, C., Barbas, C., Gonzalvez, A. L., Guttman, A., Szigeti, M., Britz-McKibbin, P., Kroezen, Z., Shanmuganathan, M., Nemes, P., Portero, E. P., Hankemeier, T., Codesido, S., Gonzalez-Ruiz, V., Rudaz, S., Ramautar, R., Anal Chem 2020, 92, 14103-14112.
6. Latosinska, A., Siwy, J., Mischak, H., Frantzi, M., Electrophoresis 2019, 40, 2294-2308.
7. Tofte, N., Lindhardt, M., Adamova, K., Bakker, S. J. L., Beige, J., Beulens, J. W. J., Birkenfeld, A. L., Currie, G., Delles, C., Dimos, I., Francova, L., Frimodt-Moller, M., Girman, P., Goke, R., Havrdova, T., Heerspink, H. J. L., Kooy, A., Laverman, G. D., Mischak, H., Navis, G., Nijpels, G., Noutsou, M., Ortiz, A., Parvanova, A., Persson, F., Petrie, J. R., Ruggenenti, P. L., Rutters, F., Rychlik, I., Siwy, J., Spasovski, G., Speeckaert, M., Trillini, M., Zurbig, P., von der Leyen, H., Rossing, P., investigators, P., Lancet Diabetes Endocrinol 2020, 8, 301-312.
8. Weissinger, E. M., Human, C., Metzger, J., Hambach, L., Wolf, D., Greinix, H. T., Dickinson, A. M., Mullen, W., Jonigk, D., Kuzmina, Z., Kreipe, H., Schweier, P., Bohm, O., Turuchanow, I., Ihlenburg-Schwarz, D., Raad, J., Durban, A., Schiemann, M., Konecke, C., Diedrich, H., Holler, E., Beutel, G., Krauter, J., Ganser, A., Stadler, M., Leukemia 2017, 31, 654-662.
9. Wendt, R., Kalbitz, S., Lubbert, C., Kellner, N., Macholz, M., Schroth, S., Ermisch, J., Latosisnka, A., Arnold, B., Mischak, H., Beige, J., Metzger, J., Proteomics 2020, e2000202.
10. Lindenburg, P. W., Haselberg, R., Rozing, G., Ramautar, R., Chromatographia 2015, 78, 367-377.
11. DiBattista, A., McIntosh, N., Lamoureux, M., Al-Dirbashi, O. Y., Chakraborty, P., Britz-McKibbin, P., J Proteome Res 2019, 18, 841-854.
12. Saoi, M., Li, A., McGlory, C., Stokes, T., von Allmen, M. T., Phillips, S. M., Britz-McKibbin, P., Metabolites 2019, 9.
13. Harada, S., Hirayama, A., Chan, Q., Kurihara, A., Fukai, K., Iida, M., Kato, S., Sugiyama, D., Kuwabara, K., Takeuchi, A., Akiyama, M., Okamura, T., Ebbels, T. M. D., Elliott, P., Tomita, M., Sato, A., Suzuki, C., Sugimoto, M., Soga, T., Takebayashi, T., PLoS One 2018, 13, e0191230.
14. Soga, T., Ohashi, Y., Ueno, Y., Naraoka, H., Tomita, M., Nishioka, T., J Proteome Res 2003, 2, 488-494.
15. Yamamoto, M., Ly, R., Gill, B., Zhu, Y., Moran-Mirabal, J., Britz-McKibbin, P., Anal Chem 2016, 88, 10710-10719.
16. Sugimoto, M., Ota, S., Kaneko, M., Enomoto, A., Soga, T., Bio Protoc 2020, 10, e3797.
17. Zhang, W., Hankemeier, T., Ramautar, R., Methods Mol Biol 2019, 1972, 165-172.
18. Zhang, W., Segers, K., Mangelings, D., Van Eeckhaut, A., Hankemeier, T., Vander Heyden, Y., Ramautar, R., Electrophoresis 2019, 40, 2309-2320.
19. Hirayama, A., Ikeda, S., Sato, A., Soga, T., Methods Mol Biol 2019, 2030, 307-313.
20. Shanmuganathan, M., Kroezen, Z., Gill, B., Azab, S., de Souza, R. J., Teo, K. K., Atkinson, S., Subbarao, P., Desai, D., Anand, S. S., Britz-McKibbin, P., Nat Protoc 2021.
21. Onjiko, R. M., Portero, E. P., Moody, S. A., Nemes, P., J Vis Exp 2017.
22. Zhang, W., Gulersonmez, M. C., Hankemeier, T., Ramautar, R., J Vis Exp 2016.
23. Shanmuganathan, M., Macklai, S., Barrenas Cardenas, C., Kroezen, Z., Kim, M., Zizek, W., Lee, H., Britz-McKibbin, P., J Vis Exp 2019.
24. Chetwynd, A. J., Zhang, W., Faserl, K., Thorn, J. A., Lynch, I., Ramautar, R., Lindner, H. H., J Vis Exp 2020.
25. Wells, S. S., Dawod, M., Kennedy, R. T., ELECTROPHORESIS 2019, 40, 2946-2953.
26. van Mever, M., Segers, K., Drouin, N., Guled, F., Heyden, Y. V., Van Eeckhaut, A., Hankemeier, T., Ramautar, R., Microchemical Journal 2020, 156.
27. Qi, M., Philip, M. C., Yang, N., Sweedler, J. V., ACS Chem. Neurosci 2018, 9, 40-50.
28. Lombard-Banek, C., Li, J., Portero, E. P., Onjiko, R. M., Singer, C. D., Plotnick, D. O., Al Shabeeb, R. Q., Nemes, P., Angew Chem Int Ed Engl 2021.
29. Kuehnbaum, N. L., Kormendi, A., Britz-Mckibbin, P., Analytical chemistry 2013, 85, 10664.
30. Macedo, A., Mathiaparanam, S., Brick, L., Keenan, K., Gonska, T., Pedder, L., Hill, S., Britz-Mckibbin, P., ACS Central Science 2017, 3, 904-913.
31. Dibattista, A., McIntosh, N., Lamoureux, M., Al-Dirbashi, O. Y., Chakraborty, P., Britz-Mckibbin, P., Analytical chemistry 2017, 89, 8112-8121.
32. Azab, S. M., de Souza, R. J., Teo, K. K., Anand, S. S., Williams, N. C., Holzschuher, J., McGlory, C., Philips, S. M., Britz-Mckibbin, P., Journal of lipid research 2020, 61, 933-944.
33. Azab, S., Ly, R., Britz-McKibbin, P., Anal Chem 2019, 91, 2329-2336.
34. Quirino, J. P., Terabe, S., Science 1998, 282, 465-468.
35. Moreno-Gonzalez, D., Torano, J. S., Gamiz-Gracia, L., Garcia-Campana, A. M., de Jong, G. J., Somsen, G. W., Electrophoresis 2013, 34, 2615-2622.
36. Prior, A., Moldovan, R. C., Crommen, J., Servais, A. C., Fillet, M., de Jong, G. J., Somsen, G. W., Anal Chim Acta 2016, 940, 150-158.





















-(1).jpg)