Liquid chromatography
Published over 9 years ago. See the latest and most current information on Liquid chromatography.
A reproducible hydrophilic interaction liquid chromatography retention model has been developed for use with a fused-core Penta-HILIC column. This model is used for the identification of procainamide tagged N-linked glycans profiles based on their composition, with unique retentions for linkage and positional isomers. A dextran ladder reference was used to experimentally determine the retention of N-linked glycans from well characterised standards in glucose units, and coefficients representing the chromatographic influence of each glycan subunit were calculated via multi-variable linear regression. A model was developed using these coefficients, by which retention for individual glycans can be accurately predicted when utilising HILIC LCMS systems. This retention model provides an effective means of identifying isomeric glycoforms when employing HILIC LCMS analysis.
Glycosylation is a form of co-translational and post-translational modification of proteins, involving an enzymatic process that results in the attachment of glycans to the proteins. N-linked glycans are attached to the nitrogen atom of an asparagine (Asn, N) residue that is part of an Asn-X-Ser/Thr consensus sequence (X can be any amino acid with the exception of proline) [1]. N-linked glycosylation is a dynamic process that allows cells to produce complex and diverse structures, which can be altered because of changes to the environment of the cell, making glycan research a challenging prospect [1]. The absolute prevalence of these glycosylated proteins in nature is not known, but protein databases currently suggest that over half of all proteins are N-glycosylated [2].
N-linked glycans fulfil several biological roles, both functional and structural, from cell signalling and interaction to protein folding and immune responses [1,3]. N-linked glycans on glycoproteins have been the subject of many epidemiological research studies, resulting in greater understanding of various diseases such as various cancers and influenza [1,4-15]. Additionally, changes in the abundance and structure of glycans have been and continue to be explored as biomarkers for different disease states [10,13-15]. The recent advances in research have served to raise awareness that the ability to ascertain more refined linkage and position information is of vital importance, as structure is proving to be of critical importance to the function of a glycan. Even the smallest of changes in linkage can produce a significant change in function, therefore it is essential that analytical methods be able to reliably characterise glycans.
Glycoproteins have arisen as promising biopharmaceutical therapeutics and diagnostic agents in recent research endeavours. Monoclonal antibodies (mAbs), for example, are used as therapeutics for several diseases. The specific structure of the N-linked glycans on these glycoproteins, including the linkage and positional isomers, are proven to have a significant impact on the effectiveness of glycoprotein therapeutics [7,11]. In light of the alterations to the efficacy of therapeutics that structural changes can have, development of reliable glycan characterisation methods is key for quality control [11,16]. Advances in research such as these have served to bring into focus the need for more sophisticated methods of glycan analysis.
A general strategy employed when analysing the structures of N-linked glycans involves releasing the glycans from the protein(s) via enzymatic or chemical digestion protocols. After the glycans are released, there are several possible methods for analysis available. A common practice consists of labelling the detached glycans with a fluorescent tag [17,19], such as procainamide (ProA) or 2-aminobenzamide (2-AB). Each of these commercially available fluorescent tags provides a number of advantages when analysing N-linked glycans [5,12,14,17-20]. Both allow for fluorescence and UV detection, straightforward quantification, and increased sample solubility in high organic systems. Since hydrophilic interaction liquid chromatography uses high concentrations of organic solvents, the ability to dissolve samples in these solvents is necessary for LC separation. The ProA fluorescent tag is shown to provide greater fluorescence signal intensity, better labelling efficiency for minor glycan species, and it also facilitates significantly enhanced electrospray ionisation efficiency. These characteristics result in an increased MS sensitivity due to the increase in ionisation and signal intensity, and an improvement in the ability to identify minor glycan species due to the increased labelling efficiency [18-19].
There are several instruments used for the MS analysis of N-linked glycans, and the one predominantly used in this research is a triple quadrupole mass spectrometer (QqQ). While the mass resolution and range is exceeded by sector instruments, the QqQ has the benefit of having superior detection sensitivity and quantification while being efficient, easy to operate, and cheaper than some other traditional MS instruments [21]. The instrument setup of the QqQ allows for four types of scans to be performed, and each has a specific purpose [21,22]. The scan type used for the development of the retention model is called a selected reaction monitoring (SRM) scan. During an SRM scan, the m/z values of the precursor ions in quadrupole 1 (Q1) and the m/z values of fragment ions in quadrupole 3 (Q3) are used. Using both of these values allows only specific fragments from specific precursor ions to be detected, which results in greater sensitivity and a lower detection limit than precursor ion scans alone. If retention information for the N-linked glycans of interest is known, it can be incorporated into the SRM method to produce a scheduled SRM (sSRM) experiment. The addition of retention information allows for fewer transitions to be observed by the instrument at any given time, thereby increasing dwell time and providing more data points across chromatographic peaks. The integration of retention information not only results in improved accuracy and reproducibility, it also allows more analytes to be included without compromising the integrity of the experiment. While the use of SRM experiments is still relatively uncommon in glycomics research, the successful use of this technique in medical and pharmaceutical industries for proteomics research and analysis is well established [4,7,12,14,23].
Using an MS instrument alone may provide the structural identification of N-linked glycans, but obtaining specific details such as linkage identification is a much more difficult prospect. Several isomers can have the same m/z but have differences in things like the linkage position, such as glycans with a 2,3-linked versus 2,6-linked sialic acid. The incorporation of an LC method using a hydrophilic interaction liquid chromatography (HILIC) column into an experiment can result in separation of isomers such as these so identification is significantly easier. As glycan profiles are experimentally obtained, a database can be constructed and used to assist in identification of glycans during subsequent experiments [24,25]. Databases like these require time to develop, can only be used once a glycan profile is known, and may not provide sufficient information for isomer identification. The prediction model described here is an effective tool designed to complement the identification of isomeric glycoforms via HILIC LC-MS analysis. The model calculates retention based upon the individual influences that individual monosaccharide species have during separation, with variations attributable to linkage and position. This predictive tool allows both knowns and unknowns to be identified more readily and with greater precision.
Acetic acid, acetonitrile, alpha-1-acid glycoprotein (bovine), alpha-1-acid glycoprotein (human), asialofetuin (bovine), dextran ladder, dimethyl sulphoxide (DMSO), fetuin (bovine), human serum (human male, AB plasma), methanol, ovalbumin, procainamide hydrochloride, ribonuclease B, and trypsin (TPCK treated) were purchased from Sigma Aldrich (St. Louis, MO, US). Ammonium bicarbonate (AMBIC), ammonium formate, formic acid, and sodium cyanoborohydride were purchased from Fluka. PNGase F (glycerol free) was purchased from New England Biolabs (Ipswich, MA, United States).
N-linked Glycan Release and Purification
A combination of ribonuclease B, ovalbumin, alpha-1-acid glycoprotein (human), alpha-1-acid glycoprotein, fetuin (bovine), and asialofetuin (bovine) was dissolved in 50 mM AMBIC, pH = 7.6. The sample solution was heated at 100°C for five minutes to denature the proteins, then allowed to cool for five minutes. Trypsin digestion was performed with an overnight incubation at 37°C with an appropriate aliquot of enzyme solution added to the sample (1 mg TPCK treated trypsin per 100 µL AMBIC buffer, protein:trypsin = 20:1). Trypsin enzyme deactivation was performed by heating the sample at 100°C for five minutes. Enzymatic release of N-linked glycans was done with an overnight incubation at 37°C with an appropriate aliquot of PNGase F added to the sample (protein:PNGase F = 1 mg:6 IUB milliunits). Released N-linked glycans were separated from the rest of the solution by reverse-phase liquid chromatography using a C18 SPE column, then frozen and lyophilised.
A modified form of Klapoetke’s procedure [18] was used to label the released N-linked glycans with ProA. Labelling solutions were freshly prepared by adding 54 mg procainamide hydrochloride and 31.5 mg sodium cyanoborohydride per 500 µL of 7:3 (v/v) dimethyl sulphoxide and acetic acid mixture. ProA labelling was performed with an overnight incubation at 37°C with 50 µL aliquots of labelling solution added to each sample. Excess labelling solution was removed from the samples using MiniTrap G-10 size exclusion columns. The ProA labelled released N-linked glycans were frozen and lyophilised, then stored until required for analysis.
Procainamide labelled N-linked glycan separation was achieved by means of a Nexera UFLC (Shimadzu) and a Halo Penta-HILIC column (2.1 mm id, 150 mm length, 2.7 µm particle size) (Advanced Materials Technology, Wilmington, DE, US). Column temperature was set to 60°C, and the solvent flow rate was 0.4 mL/min. Mobile phase A was a solution of 50 mM ammonium formate, 5% acetonitrile, and 0.1% formic acid in water and mobile phase B was acetonitrile. A linear gradient of 78%-48% mobile phase B in 75 minutes was used for all experiments. This gradient provided adequate separation for the wide variety of glycoforms in both simple and complex samples while expediting complete elution with minimal undesirable effects to either the experimental parameters or the resulting data. Samples were dissolved in 78% acetonitrile before injection in the UFLC system.
Selected reaction monitoring (SRM) and scheduled SRM experiments were conducted on a hybrid quadrupole/linear ion trap mass spectrometer (4000 QqQ, Applied Biosystems/MDS SCIEX, Foster City, CA, United States) using an electrospray source. N-linked glycan ion (Q1) values for each standard were calculated using GlycoWorkbench software, and a fragment ion (Q3) value of m/z 440.8 representing ProA and GlcNAc was used.
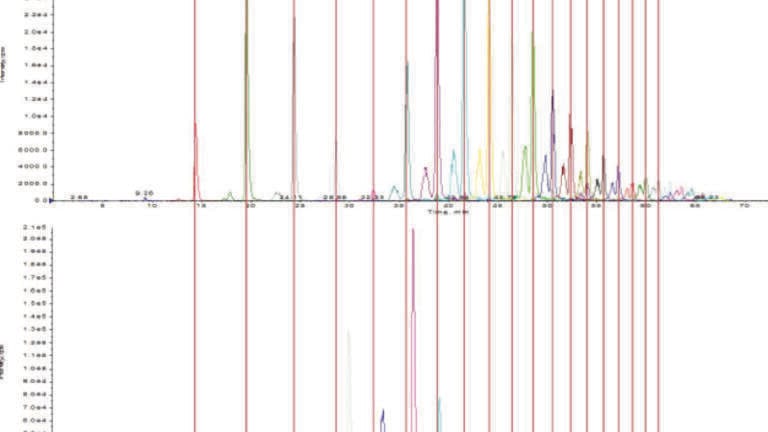
The standard glycoproteins used in this study were selected because they have well-characterised glycans. These standards also have a diverse array of glycoform structures, including high mannose, complex, and hybrid structures. Dextran ladder is also well known, commonly used as a reference standard, and was used to correlate time in minutes to glucose units. LC-MS analysis was performed on the released N-linked glycans from the combined glycoprotein standards, and 92 N-linked glycan signals were selected for analysis. The retention time in minutes for each glycan selected was converted into glucose units using the dextran ladder standard (Figure 1). After conversion of retention in minutes to glucose units was completed for each glycan, the amounts of the chromatographically influencing monosaccharides that comprise each glycan were determined, with separate values for applicable variations in linkage and position. Using the retention information in glucose units and the number of each monosaccharide species present in the glycans, a multi-variable linear regression analysis was performed. The objective of this form of analysis is to produce a model that represents the relationship between two or more independent explanatory variables (x), in this case the individual monosaccharide species, and a dependent response variable (y), in this case retention in glucose units, by fitting a linear equation to the experimental data. The model for multiple linear regression, given i observations, is written as:
yi = β0+ β1 x i,1 + β2 xi,2+ … + βp xi,p+ ∈i
Using this linear regression model and one of several solving methods, a coefficient (β) for each x value can be calculated. (Table 1) The coefficient is the average change in the response variable for one unit of change in the predictor variable while all other model predictors are kept constant. Various relevant statistical values can also be calculated for each coefficient, including standard error, t-stat, P-value, and upper/lower 95% confidence values, also shown in Table 1. The standard error is an estimation of the standard deviation of the coefficient, and is used as a measure of how precise the coefficient measurement is. All coefficients have a standard error below 0.2 and all but one have a standard error below 0.1, indicating the calculated coefficients show little variation across different cases. The t-stat is calculated by dividing the coefficient by the standard error. If the sample size exceeds 30 observations, as is the case with this work, then a t-stat great than 2 or less than -2 is an indication that the coefficient is significant with >95% confidence. The calculated t-stat values are well outside the -2/2 range, indicating the coefficients are very likely to be statistically significant with a high level of confidence. The p-value is used to test the null hypothesis, and a p-value lower than 0.05 indicates that the null hypothesis may be rejected. To state this in a different way, if a coefficient has a sufficiently low p-value, changes in the predictor value are associated with changes in the response variable. If the p-value is larger, then changes in the predictor value are not associated with changes in the response variable, therefore that value should be removed and another fit attempted. All calculated p-values were well below the threshold necessary to reject the null hypothesis; therefore, all coefficients are meaningful contributions to the prediction model. The upper and lower 95% confidence level thresholds represent the range in which there is a 95% probability the coefficient lies, which means there is only a 2.5% chance that it would be above the upper value or below the lower value, and the narrower this range is the better. All upper and lower ranges were <1, again showcasing the precision of the predictor.
Once the coefficients are calculated, retention values in glucose units for N-linked glycans can be calculated using the retention model equation:
R = ∑ NxMx+b
(Nx is the number of a particular monosaccharide x present in the glycan of interest, Mx is the coefficient of monosaccharide x, b is the intercept of the retention model). Retention is calculated in glucose units, which can be correlated to minutes by way of the dextran ladder standard, so this model may easily be used with other LC-MS systems.
Using the retention model equation, prediction values for the N-linked glycan standards were calculated and compared to experimental values. A graph of calculated versus experimental glucose units was constructed (Figure 2), and the linear trend line of this graph displayed a high correlation coefficient (R2 = 0.9941). Standard deviation in minutes for calculated versus experimental time was calculated by converting the glucose unit values to minutes by way of the dextran ladder reference. These calculations revealed a standard deviation of ≤ ±2 minutes. The high level of correlation and linear relationship between the calculated and experimental values exhibits the effectiveness of the retention prediction model.
The ability to predict retention for glycans assists analysis in two major ways. First, it provides the retention information for glycans that have not been observed previously. This information allows for the scheduling of selected reaction monitoring (SRM) transitions for these glycans, enhancing detection and confirming identifications during the experiment. Second, predicting retention can assist in identifying glycans subsequent to LCMS analysis. Searching software databases often results in either erroneous or incomplete identification of the observed glycans, therefore a retention model can be used to select an identity out of several possibilities or define specific details of an identified glycan. An example of this is shown in Figure 3, where the four isomers of two closely related sialic acid species are identified. Both are singly sialylated biantennary N-linked glycans, with one trace representing the α2,3- and α2,6-linked NeuAc isomers and the other representing the α2,3- and α2,6-linked NeuGc isomers. While the m/z values can distinguish which pair contains NeuAc and which pair contains NeuGc, it cannot distinguish between linkage isomers as there is no m/z difference between them. Using the retention model, it is a simple matter to assign specific linkage details to the two signals of either moiety. The versatility of the model, having been designed in such a way as to be of use with variations to experimental parameters, makes it a valuable tool for analytical procedures, both prior and subsequent to LC-MS experiments.
The use of glucose units instead of minutes for the retention model allows for its use on different instruments or with experimental parameter modifications while still maintaining prediction accuracy. As instruments can experience retention shifts over time, and since different instruments intrinsically have variables such as void volume, adaptability for retention calculations is essential. A simple experiment with a dextran ladder standard can easily provide the necessary information for either predicting retention times in a scheduled experiment or for aiding in analyte identification in a completed experiment. Another key aspect of the model as it has been constructed which enhances its versatility is that additional coefficients can easily be incorporated, which will expand its application.
Support for this work comes from NIH grant GM0 93747 to B.B.
1. A. Varki, R. Cummings, J. Esko, H. Freeze, P. Stanley, C. Bertozzi, G. Hart, M. Etzler. Essentials of Glycobiology. 2nd ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, 2009.
2. R. Apweiler Biochim. Biophys. Acta, Gen. Subj. 1473 (1999) 4-8.
3. K. D. Maureen E. Taylor. Introduction to Glycobiology. 2nd ed.; Oxford University Press: UNITED STATES, 2006.
4. Y. H. Ahn, P. M. Shin, N. R. Oh, G. W. Park, H. Kim, J. S. Yoo J. Proteomics 75 (2012) 5507-5515.
5. F. Higel, U. Demelbauer, A. Seidl, W. Friess, F. Sorgel Anal. Bioanal. Chem. 405 (2013) 2481-93.
6. T. Ito, Y. Suzuki, T. Suzuki, A. Takada, T. Horimoto, K. Wells, H. Kida, K. Otsuki, M. Kiso, H. Ishida, Y. Kawaoka J. Virol. 74 (2000) 9300-9305.
7. J. W. Lee, D. Figeys, J. Vasilescu Adv. Cancer Res. 96 (2006) 269-298.
8. I. Loke, N. H. Packer, M. Thaysen-Andersen Biomolecules 5 (2015) 1832-54.
9. M. Matrosovich, A. Tuzikov, N. Bovin, A. Gambaryan, A. Klimov, M. R. Castrucci, I. Donatelli, Y. Kawaoka J. Virol. 74 (2000) 8502-8512.
10. E. Miyoshi, K. Moriwaki, N. Terao, C. C. Tan, M. Terao, T. Nakagawa, H. Matsumoto, S. Shinzaki, Y. Kamada Biomolecules 2 (2012) 34-45.
11. T. Mizushima, H. Yagi, E. Takemoto, M. Shibata-Koyama, Y. Isoda, S. Iida, K. Masuda, M. Satoh, K. Kato Genes Cells 16 (2011) 1071-80.
12. T. Nishimura, M. Nomura, H. Tojo, H. Hamasaki, T. Fukuda, K. Fujii, S. Mikami, Y. Bando, H. Kato J. Proteomics 73 (2010) 1100-10.
13. Y. Suzuki, T. Ito, T. Suzuki, R. E. Holland, T. M. Chambers, M. Kiso, H. Ishida, Y. Kawaoka J. Virol. 74 (2000) 11825-11831.
14. M. Y. Xu, Y. Qu, X. F. Jia, M. L. Wang, H. Liu, X. P. Wang, L. J. Zhang, L. G. Lu Biomed Pharmacother 67 (2013) 561-7.
15. Y. P. Zhao, X. Y. Xu, M. Fang, H. Wang, Q. You, C. H. Yi, J. Ji, X. Gu, P. T. Zhou, C. Cheng, C. F. Gao PLoS One 9 (2014) e94536.
16. N. Yamane-Ohnuki, M. Satoh mAbs 1 (2014) 230-236.
17. J. Ahn, J. Bones, Y. Q. Yu, P. M. Rudd, M. Gilar J. Chromatogr., B: Anal. Technol. Biomed. Life Sci. 878 (2010) 403-8.
18. S. Klapoetke, J. Zhang, S. Becht, X. Gu, X. Ding J. Pharm. Biomed. Anal. 53 (2010) 315-24.
19. R. P. Kozak, C. B. Tortosa, D. L. Fernandes, D. I. Spencer Anal. Biochem. 486 (2015) 38-40.
20. G. Zauner, C. A. Koeleman, A. M. Deelder, M. Wuhrer J. Sep. Sci. 33 (2010) 903-10.
21. C. Dass. Fundamentals of Contemporary Mass Spectrometry. John Wiley and Sons, Inc.: Hoboken, NJ, 2007.
22. E. de Hoffmann J. Mass Spectrom. 31 (1996) 129-137.
23. C. E. Parker, T. W. Pearson, N. L. Anderson, C. H. Borchers Analyst 135 (2010) 1830-8.
24. P. Pompach, K. B. Chandler, R. Lan, N. Edwards, R. Goldman J. Proteome Res. 11 (2012) 1728-40.
25. L. Royle, M. P. Campbell, C. M. Radcliffe, D. M. White, D. J. Harvey, J. L. Abrahams, Y. G. Kim, G. W. Henry, N. A. Shadick, M. E. Weinblatt, D. M. Lee, P. M. Rudd, R. A. Dwek Anal. Biochem. 376 (2008) 1-12.























-(1).jpg)