Ion chromatography (IC)
Published over 5 years ago. See the latest and most current information on Ion chromatography (IC).
Small organic acids are used throughout the pharmaceutical industry [1], but are challenging analytically when using conventional methods such as reverse-phase high performance liquid chromatography (HPLC) and gas chromatography (GC). Ion chromatography (IC) offers better analytical separation of these acids, but conductivity detection does not provide peak purity information. When coupled with mass spectrometry (MS), information about components contributing to each peak is obtained. IC-MS was used for the method development of 2-butynoic acid impurity analysis; where it was possible to analyse the impurities and confirm their peak purity. Quantitative performance of the method for sensitivity and linearity for a series of organic acids, spiked into 2-butynoic acid at levels representative of impurities, was evaluated [2].
Organic acids are used as reagents in pharmaceutical synthetic processes or potentially formed as impurities. They need to be controlled throughout the synthesis and manufacturing routes to ensure a high-quality final product. Trace impurities present in starting or intermediate materials, if undetected, can directly impact the quality of the final product. Discovering discrepancies in the product quality at later stages makes it technically challenging and expensive to identify contaminants across different preceding stages of the production process. The purer the starting materials and intermediates, the easier it is to control and assure the quality of the final product. Accurate and reliable analytical detection of organic acid impurities is, therefore, an extremely important requirement for those who manufacture products for internal use. It is essential to have methods that can identify and quantify all possible impurities when investing in expensive supply of starting material or intermediates.
The widely used analytical methods, gas chromatography (GC) and high performance liquid chromatography (HPLC), when applied towards organic acid analyses face numerous challenges [3,4]:
• Small organic acids, many of which are highly polar molecules, are difficult to retain on reversed-phase LC columns.
• As they are often poor chromophores, using UV detection to analyse small organic acids can produce varying response factors and poor sensitivity such that this cannot be used for accurate quantitation, especially at lower levels.
• Performing measurements with detection at lower wavelengths also poses some restrictions on the type of solvents or additives used as eluent components, further limiting the scope of LC analyses.
• For samples to be analysed using GC, they often need to be derivatised, adding an extra level of sample handling [5,6], and consequently making the protocol more time-consuming.
Ion chromatography (IC) with suppressed conductivity detection (sCD), on the other hand, is a promising method to analyse organic acids without derivatisation. When coupled with MS, it offers better sensitivity and higher resolution of potential analytes [7,8], enabling accurate quantitation of resolved peaks and improved quality assurance. Ion chromatography takes advantage of the fact that organic acids are readily charged through the addition of a basic additive to the eluent. The separation of the acids is gained through ion-exchange of the negatively charged acidic group with the ammonium resin on the column with a basic eluent [9].
IC is inherently better suited for analysing polar molecules: the separation is highly dependent on the analyte’s pKa and size-to-charge ratio. However, IC analysis with conductivity detection does not offer absolute identification of the components corresponding to each peak, in turn, not allowing the accurate quantitation of all trace impurities present if they partially or fully coelute. Consequently, as peak purity tracking is not directly possible with just IC-sCD, it hinders peak confirmation for identifying unknowns. Despite its promise, IC methods, therefore, have been underutilised in organic acid analysis.
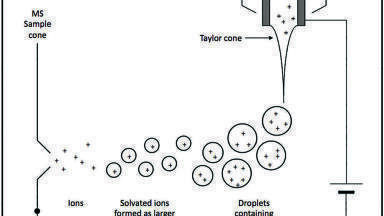
One way to improve the overall selectivity is to couple IC with MS. The mass information obtained for each peak allows another level of resolution of the corresponding components, increasing the specificity and selectivity of the analytical approach. However, the strong basicity of eluents used in anionic analysis by IC are incompatible with electrospray ionisation (ESI) MS due to their possible corrosive effects on the stainless steel components of the MS ion source, adding another challenge to overcome. In recent times, eluent suppressors have made MS coupling easier [9,10] as they suppress the eluent by removing the counter ions before introducing them into the MS ion source as water, thereby enabling ionisation of the analyte to happen.
Hyphenating IC with MS along with eluent suppressors can address the lack of obtainable information on peak purity. This IC-MS approach to analyse polar organic acids, when appropriately developed and optimised, can serve as a superior alternative to HPLC and GC methods in terms of sensitivity and robustness. In addition to sensitive measurements, quality control and quality assurance laboratories across different industries need reliable and reproducible methods, with quantitative capabilities in the required dynamic range, to routinely detect organic acid impurities to minimise risks upfront.
Here, we’ve developed a method for purity analysis of 2-butynoic acid, along with trace level impurity identification, to facilitate successful synthetic processes in the pharmaceutical industry. 2-butynoic acid, containing a weak chromophore, typically yields poor sensitivity in HPLC-UV analytical measurements, making it a good candidate to validate the robustness of IC-MS. In this method, anionic separation coupled with ESI MS employing negative ionisation was used to analyse impurities present in 2-butynoic acid samples. Limits of quantification and linearity of detection by both IC-sCD and MS were evaluated. The developed method was then used to study a range of organic acids potentially present as impurities at pharmaceutically relevant levels.
Standards, materials and instruments
Standards of 2-butynoic acid and other organic acids (namely, acetic acid, propinoic acid, formic acid, butanoic acid, crotonic acid, pentanoic acid, propiolic acid, pentynoic acid) were obtained from Sigma-Aldrich Company Ltd (Gillingham, United Kingdom). Samples were diluted with 18.2MΩ-cm ultrapure water obtained from a MilliQ unit (Watford, Hertfordshire). HPLC grade 0.25 M ammonia and 0.25 M ammonium acetate were obtained from Fisher Scientific (Loughborough, UK). Samples of 2-butynoic acid were obtained from FAR Chemical (Florida, USA), Boropharm (Michigan, USA) and AstraZeneca (Macclesfield, UK).
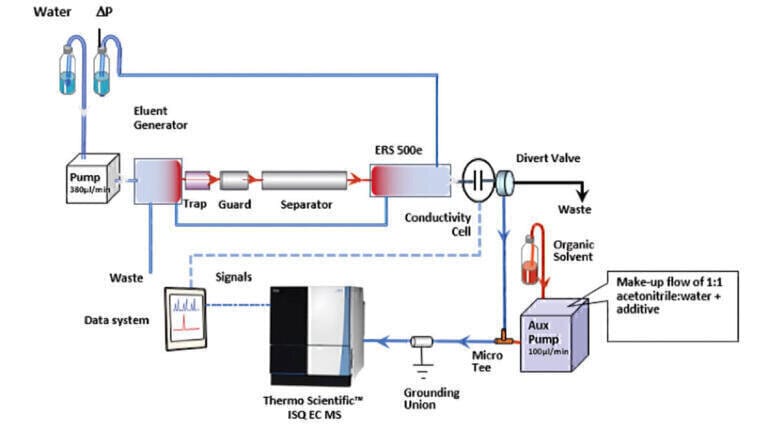
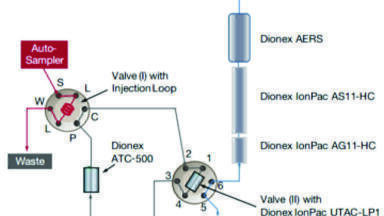
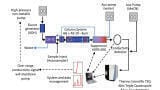
IC-MS was performed on a Thermo Scientific Dionex Integrion High Pressure Ion Chromatograph (Hemel Hempstead, UK) coupled to an ISQ EC mass spectrometer (Thermo Fisher Scientific, Hemel Hempstead, UK). The IC-MS workflow was set up as shown in Figure 1.
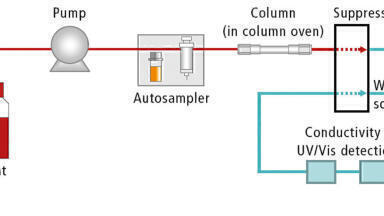
The IC systems comprised of a guard column (Dionex IonPac™ AG11-HC-4 μm 2 ×50 mm), an IC column (Dionex IonPac™ AS11-HC-4 μm 2 ×250 mm) and suppressor (initially, Dionex AERS 500e 2 mm and then changed to Dionex ADRS 600 2 mm) with conductivity detector. The Dionex AERS 500e suppressor applies a direct fixed current to suppress the hydroxide to water, and the potassium counter-ions are vented to waste as they are pulled through the exchange membrane. A suppressor current of 51 mA was used unless otherwise stated. Dionex ADRS 600 suppressor applies a direct fixed voltage to give the most suitable current to match the concentration of the eluent to suppress the hydroxide to water. A voltage of 4 V was used. The flow rate was maintained at 0.38 mL/min and an injection volume of 25 μL was used for all injections. The specifications of the isocratic and gradient methods are as follows:
• Isocratic method: 30 mM KOH, 30 °C, lower current 29 mA
• Gradient method: Initial 1 mM KOH for 8.5 min, then linear gradient to 15 mM over 10 min, then to 30 mM over a further 10 min and then to 54 mM in 1.5 min.
MS conditions
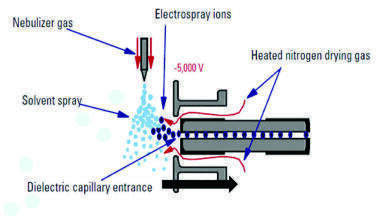
All MS experiments were performed using the following conditions: vaporiser temperature of 450°C, ion transfer tube temperature of 200°C, ionisation voltage of -2500 V, sheath gas pressure of 50 psi and auxiliary gas pressure of 5 psi. The system was operated in negative ion ESI mode for all analyses. The MS system, when operated in full scan mode, recorded data for the mass range of m/z 40-250. Selected Ion Monitoring (SIM) mode was used for the analysis of deprotonated molecules. The MS ion source voltage (applied to aid desolvation) was set to 5 V for both the full scan and SIM analyses. Make-up flow of 1:1 acetonitrile: water (+ buffer additive, if used) was added at a rate of 0.1 mL/min.
Standards and sample preparation
Individual organic acid stock standard solutions were prepared at 10 ppm (v/v) or 10 mg/L, except for butynoic, crotonic, and pentynoic acid solutions which were prepared at 50 ppm (v/v) or 50 mg/L. Mixed working stock solutions of 1 ppm and 100 ppb were prepared by diluting the stock solutions 10- and 100-fold respectively (50- and 500-fold for 50 ppm stock). These working stock solutions were then diluted to prepare a range of calibration standards between 1 ppb and 500 ppb. The 1 ppm working solution was used to prepare the 500 ppb, 200 ppb, and 100 ppb standards, and the 100 ppb working solution was used to prepare the 50 ppb, 10 ppb, 5 ppb, and 1 ppb standards.
Results and discussion
Adding ammonia to the make-up flow provided the strongest m/z response
The main impurity of 2-butynoic acid, analysed using the isocratic method, produced an ion with m/z 127 with a partially co-eluting impurity at m/z 171 as shown by the extracted ion chromatograms (Figure 2A). Using different additives in the make-up flow resulted in different MS signal responses as shown in Figure 2. Based on its chemical composition, each additive (MeCN: H2O, MeCN: H2O + 12.5 mM ammonium acetate and MeCN: H2O + 25 mM ammonia) helps to volatilise the analytes in solution and, in turn, contributes to the efficiency of the ionisation process. Of these additives, using 25mM ammonia in the make-up solvent provided a greater response for both m/z 127 and m/z 171 ions (Figure 2). Ammonia was therefore chosen for all subsequent experiments. As expected, the gradient method provided a better separation of peaks, thereby, offering a greater resolution of components than the isocratic method. The IC-sCD chromatogram and the extracted ion chromatograms of m/z 127 and m/z 171 are shown in Figure 3.
Using the gradient method, the separation of a range of low molecular weight organic standards that could potentially be present as impurities in 2-butynoic acid was investigated. The temperature of the column in IC has previously been reported to influence peak shape in ion-exchange chromatography. Here, the organic acid standards were analysed at column temperatures of 30, 40, 50 & 60°C. The acquired IC-sCD chromatograms corresponding to the different column temperatures are shown in Figure 4. Increasing column temperatures improved the peak shape and reduced peak tailing but resulted in losing resolution on early eluting peaks.
MS detection offered greater selectivity over CD
The organic acid standards, ranging in concentration between 1 ppb and 500 ppb, were analysed using the gradient method and the linearity was evaluated by calculating the R2 values. Table 1 shows the limit of quantitation (LOQ) and determined linearity for each of the individual organic acid components using both IC-sCD and IC-MS in the order of elution. The R2 values for the concentration range of 1 to 500 ppb were greater than 0.99 from IC-sCD for most of the acids and greater than 0.95 from the MS. With the MS method for formic acid and propiolic acid, the R2 could be increased to 0.99 by removing the highest concentration standard when assessing linearity, but that would mean the method’s linearity is only better in the lower concentration range.
The 1 ppb standard was easily detectable for each component with a signal-to-noise ratio of a minimum of 4:1, indicating that the LOQ was less than 1 ppb, apart from pentynoic acid, which had a LOQ of 5 ppb. MS response, as seen in Figure 5, had a greater signal-to-noise ratio than the IC-sCD response and did not have issues with the baseline observed in the IC-sCD chromatogram. Propiolic acid and butynoic acid could not be analysed for linearity or LOQ by IC-sCD in a mixed solution using this method due to partial co-elution. However, these closely eluting analytes differed in their masses, making it feasible to obtain resolution using MS detection, resulting in a LOQ of 1 ppb as shown by the extracted ion chromatogram (EIC) in Figure 5. The additional selectivity of MS detection after IC allowed rapid analysis of peak purity in organic acids and provided quantitation for even those impurities that were only partially separated by IC. Increased sensitivity in impurity detection makes it possible to assess known and unknown impurities more accurately during the early stages of the production processes, reducing the risk of expensive troubleshooting during later stages.
After obtaining successful linearity and LOQ for all the organic acid standards at low concentrations, it was necessary to confirm whether the linearity and LOQ of these impurities could be achieved in the presence of a 2-butynoic acid sample matrix. Table 2 shows the LOQ and linearity values for each of the individual organic acid components using both IC-sCD and IC-MS in the presence of 25 mg/L 2-butynoic acid. Linearity of response was maintained even in the presence of the 2-butynoic acid matrix. The LOQ obtained using IC-sCD was below the quantification and identification thresholds for impurities as recommended by the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) [11], making this technique suitable for routine analysis of all fully resolved impurities. MS, with even lower levels of detection and better sensitivity, can be used for trace analysis.
Relative standard deviation for results across IC with conductivity and MS detection of all organic acids within a 2-butynoic acid sample showed good reproducibility
Impurity testing in the pharmaceutical industry needs to be reliable and reproducible. The reproducibility of the IC-MS response was evaluated across ten injections of both the 50 ppb mixed organic acid samples and the 0.1%(w/w) impurity level organic acid mix in 25 mg/L of 2-butynoic acid. The relative standard deviation (RSD) was calculated from the obtained peak area values from ten injections. Using IC-sCD, all components, except for pentynoic acid, produced a peak area RSD of less than 2%, indicating excellent reproducibility. MS detection produced a peak area RSD in the range of 4-10% for the two sample types (Table 3). This MS response variability is a direct result of the underlying ionisation process but fell within the acceptable RSD range for trace analysis. These results show that even with little optimisation of MS conditions, IC-MS is reproducible across all components for routine and trace analysis.
By coupling IC with MS, it was possible to accurately quantify impurities of small organic acids such as 2-butynoic acid without derivatisation but with the benefit of absolute identification enabled by the mass spectrometric data. With linearity, LOQ, and repeatability measurements within the pharmaceutically acceptable range, this method can now be applied for routine analysis of impurities (using IC-sCD after the appropriate method development) or trace analysis (using IC-MS to leverage component identification). Additive choice for make-up solvent, a key variable in IC-MS methods, as expected, directly impacted the MS response. Optimising the column temperature allowed improvement in peak shapes.
The additional selectivity provided by MS allowed peak purity tracking measurements that were otherwise challenging with IC-sCD only. For instance, co-eluted analytes, 2-butynoic acid and propiolic acid, were not resolved in isocratic or gradient IC methods but were easily detected, resolved and quantified using extracted ion chromatograms. When faced with co-eluting impurities, rather than relying on the information obtained by their elution profiles, using information provided by measuring their masses, and extracting the corresponding ion chromatograms, allows resolving these impurities even if that is chromatographically not possible. MS hyphenation, therefore, becomes particularly important when quantifying closely eluting or overlapping impurities.
Without the need for derivatisation, IC-MS is significantly more straightforward and faster to perform, saving valuable time during method development and follow-up routine analysis. It offers advantages over reversed-phase HPLC and GC to yield sensitive and reproducible measurements of polar organic acid impurities, that can also be applied to study other polar ionic analytes such as amines and glyphosate [12].
1. Global organic acid market: Development in pharmaceutical industry stokes growth, Transparency Market Research (2018)
2. T.A. Corry, B.A. Jackson, A.D. Ray, Impurity analysis of 2-butynoic acid by ion chromatography-mass spectrometry. Journal of Chromatography. A. (2019) 460470
3. J. Št’ávová, J. Beránek, E.P. Nelson, B.A. Diep, A. Kubátová, Limits of detections for the determination of mono- and dicarboxylic acids using gas and liquid chromatographic methods coupled with mass spectrometry, J. Chromatogr. B 879 (2011) 1429–1438
4. Y. Lu, D. Yao, Chi Chen, C 2-hydrazinoquinoline as a derivatization agent for LC–MS-based metabolomic investigation of diabetic ketoacidosis, Metabolites 3 (2013) 993–1010
5. I. Molnár-Perl, Role of chromatography in the analysis of sugars, carboxylic acids and amino acids in food, J. Chromatogr. A 891 (2000) 1–32
6. M.A. Adams, Z.L. Chen, P. Landman, T.D. Colmer, Simultaneous determination by capillary gas chromatography of organic acids, sugars, and sugar alcohols in plant tissue extracts as their trimethylsilyl derivatives, Anal. Biochem. 266 (1999) 77–84
7. C. Bruggink, J. Trick, C. Wanner, D. Jensen, Novel determination of organic acids in diesel and motor oil by ion chromatography, Anal. Lett. 50 (2016) 739–747
8. X. Geng, S. Zhang, Q. Wang, Z.K. Zhao, Determination of organic acids in the presence of inorganic anions by ion chromatography with suppressed conductivity detection, J. Chromatogr. A 1192 (2008) 187–190
9. H.H. Willard, L.L. Merritt Jr., J.A. Dean, & F.A. Settle Jr. (1988). Instrumental methods of analysis, 7th edition. United States: Wadsworth Publishing Company.
10. M. C. Jin, X.-H. Chen, M.-Q. Cai, Y. Zhu, Eluent generator reagent free ion chromatography with electrospray ionization mass spectrometry for simultaneous analysis of organic acids in juices and beverages, Anal. Lett. 43 (2010) 2061–2077
11. ICH Q2 (R1) Validation of analytical procedures: text and methodology, 1994
12. Z. Lewis, B.A. Jackson, A. Crampton, A.D. Ray, S.W. Holman, Towards a generic method for ion chromatography/mass spectrometry of low-molecular-weight amines in pharmaceutical drug discovery and development, Rapid Communications in Mass Spectrometry (2020) e8680


















-(1).jpg)