Ion chromatography (IC)
Published over 7 years ago. See the latest and most current information on Ion chromatography (IC).
One of the challenges of an analytical scientist is to ensure that the developed assays are robust, and so analysing the same sample should give the same result. In this article we will look at how the use of electrospray can result in a large variability in the detector response, and how the root cause of this variability is associated with the sample preparation and the chromatography.
The advent of atmospheric pressure ionizsation (API) sources allows the coupling of high performance liquid chromatography (HPLC) to mass spectrometry (MS) [1], which has resulted in assays having lower detection limits and a greater degree of specificity. Other detector types also use an electrospray type interface such as Evaporative Light Scattering Detector (ELSD) and Charged Aerosol Detector (CAD). Electrospray ionisation (ESI) is one of the most widely used LC/MS interfaces and has applications in the analyses of a variety of different analytes such as characterisation of peptides and proteins [2,3,4], screening of drugs and steroids [5,6,7] and detection and quantification of residual pesticides in food [6,8,9,10]. The broad range of applications is due to the compatibility of the technology to a wide range of organic molecules.
Within the field of bioanalysis the use of mass spectrometry coupled to liquid chromatography has become mainstream. The initial introduction of this technology was seen as resolving many, if not all of the issues that were associated with the analysis of compounds from a biological fluid. Approaches to sample preparation were significantly simplified, with terminology such as “‘dilute and shoot” ’ being introduced into the analytical scientist vocabulary. However it was soon found that this approach also came with its own challenges which need to be addressed, notably;
• Robustness of the system
Blocked columns
Blocked syringes
Precipitation of the
sample causing blockages
• Carryover
• Ion suppression
• Sensitivity issues
The addition of sample preparation and a chromatographic separation to the work flow ensures that all of these issues are addressed to a greater or lesser extent depending on the quality of the separation and also the sample preparation that is utilised.
The sample preparation is usually the most important aspect of the whole workflow, however typically it will involve the least amount of effort to optimise this part of the process. Sample preparation is often seen as something where generic approaches are employed and does not require the level of skill that is associated with the operation of the detection and chromatographic part of the process. However, since the analytical technique is dependent on the quality of the sample that is being analysed, any deficiencies that occur within the sample preparation will be transferred into the final analysis.
The sample preparation starts with the collection of the sample to ensure that the sample is representative of what is being measured. In most cases within a bioanalytical laboratory, the sample will be a biological fluid, which means that any sampling is highly likely to be representative due to the nature of the fluid flow within a biological system.
Subsequent to the sampling, the sample is then stored, and the nature of the storage container needs to be considered carefully to ensure that sample is not lost or modified. This is particularly important for larger molecules where the possible interactions is substantially increased compared to smaller molecules where the possible interactions may be limited due to the size of the molecule. It should also be noted that the configurational arrangement of a large molecule can be influenced by the storage vessel, thus proteins can denature in the presence of a hydrophobic surface and so consequently it is important to understand the nature of the analyte and also perform a series of simple experiments to determine if there are any issues at this stage of the sample preparation. These experiments would be as simple as monitoring the concentration levels over a period of time and also monitoring in different storage vessels to determine if there is any temporal differences in the observed concentration.
Once the sample storage has been addressed, the next aspect of the sample preparation to consider is how to remove the matrix components. Typically for a biological fluid this will focus on the removal of proteinaceous components and also looking at endogenous salts etc.
The final stage of the workflow is the combination of the chromatography and detection techniques. This article will focus on the LC-MS as matrix effects do cause significant effects using this technology. The impact that the initial sample preparation has on the sample analysis can be very subtle and in certain situations will not be easily identified.
Typically, the separation is optimised for the analyte molecules and this can result in some interesting issues that would not occur when using less selective detection technology, where co-eluting peaks can be clearly seen, and late eluting peaks can also be readily seen. This results in a degree of variability in the detector response due to suppression issues and varying retention times of matrix components potentially impacting the signal observed from the same sample.
In LC-MS there is an assumption that the injected sample is fully eluted from the column, or is irreversibly retained on the column, however when dealing with very complex samples this assumption may not be correct, in particular if only a few of the components that are injected are being monitored. This is the scenario in many bioanalytical assays where the mass spectrometer is used to add further resolving power to the chromatographic separation by only monitoring for the analytes of interest. Unlike when using a less discriminative detector, matrix components that elute from the column will typically not be monitored, and thus it may not be feasible to determine if the peaks elute over one gradient cycle or require more gradient cycles to fully elute the matrix components form the column. This will result in variable amounts of matrix components being eluted dependant on the injection number within a particular batch.
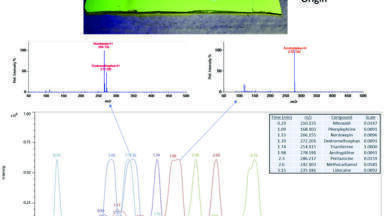
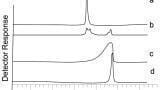
Figures 1 and 2 looks at the elution of a series of phospholipids. Figure 1 shows the chromatography and responses obtained using different sample preparation techniques. It can be seen that from Figure 1 the peak shape is poor, and that a reasonable amount of retention time space is covered by just three matrix components. Altering the sample preparation also has a significant impact in the amount of phospholipids that are left on the column, with a protein precipitation approach providing the worst approach to removing this particular matrix component. Figure 2 investigates how effectively the phospholipids are eluted from a column. The first injection is from a protein precipitated sample and it can be seen that there is a high amount of the lower molecular mass (and lower hydrophobicities) phospholipids. All subsequent injections are of water, and so any peaks that are eluting are due to system carryover. In this case it is very evident that the chromatography has not been optimised for the matrix components and this results in the phospholipids being eluted from the column over many injections. In a standard batch the amount of matrix components in different samples can vary substantially, particularly if a less effective sample preparation technique is employed. This results in potentially a very complex situation for the amount of matrix components that are eluting into the detector, as it will be dependent not only on the on the original sample, but also on the history of the column.
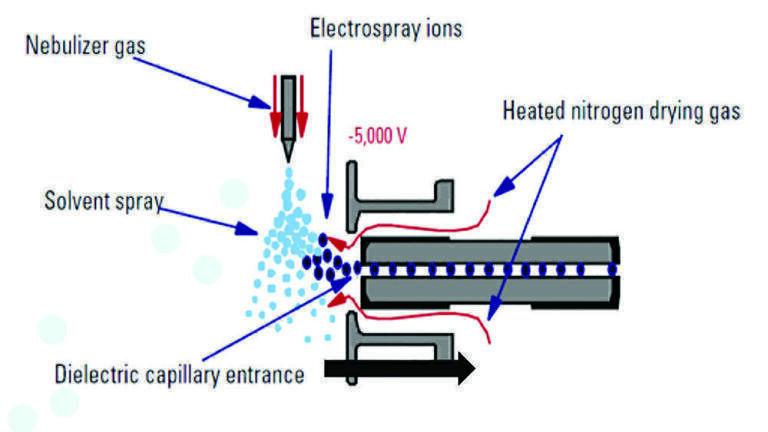
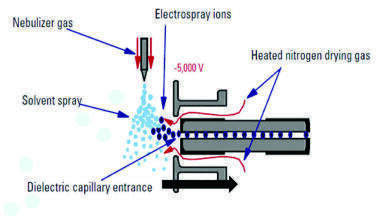
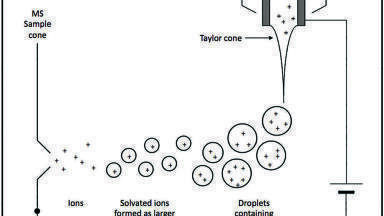
The issue with the variable matrix components is that each matrix component has the potential to interfere with the ionisation process, due to the nature of how an ESI source works. Figure 3 shows a schematic of an ESI source. An ESI source is designed such that the HPLC eluent travels through a spray capillary surrounded by a co-axial flow of nebulising gas, typically heated nitrogen. The liquid is charged by applying an electric field to the capillary, causing a potential difference between the capillary tip and the entrance of the high vacuum region of the MS. The application of a charge results in the generation of a Taylor cone. The surface tension of the eluent holds the liquid in a steady state for a set distance and beyond this an aerosol of charged droplets is formed and ejected towards the electrode of the opposing charge. Currently, there are two generally agreed mechanisms by which ions are formed from the charged liquid droplets: (a) the charged residue model (CRM) [11] ; and (b) the ion evaporation model (IEM) [12]. Both mechanisms have been demonstrated to exist together under the same experimental conditions and depending on the exact LC and MS parameters, one of these mechanisms will tend to be favoured. CRM states that solvent evaporation, often assisted by heat, causes the droplets to diminish in size, reaching a critical diameter or Rayleigh limit. At this point the residual charge on the remaining solvent is no longer stable on the available surface area of the droplet. This results in a Coulombic explosion which ideally would lead to the formation of a single molecule of analyte with a single charge associated with it. IEM proposes that droplets and ions are emitted from a much larger droplet and that this results in a gradual reduction of the size of the main droplet [11,12]. Other models do exist which explain the phenomena of electrospray [13,14,15], but these are generally variants or combinations of the CRM and IEM.
The CRM and IEM form a solid basis for understanding the processes that occur within an ionisation chamber in a mass spectrometer. These models have been further developed to allow a better understanding of the competitive ionisation process that occurs in the presence of co-eluting components. The models also refine some of the original hypothesis of the underlying processes that are occurring. In particular the ion evaporation model has seen development to allow a better comparison with experimental data. Tang and Kebarle [16] further developed the Iribane and Thompson model proposing a model that related the ion evaporation rate to the concentration of the ion in the droplet. This model was further refined by Enke [17] who proposed using the ratio of equilibrium partitioning constants rather than the rate approach. The theory proposed by Enke looks at the competitive nature of the evaporation of the ions from the droplet state into the gas phase. This process is dependent on the concentration of ions at the surface of the droplet, which can be defined as follows;
Where
Similarly
Representing the equilibrium of the analyte and electrolyte ions between the surface and interior of the droplet and;
[A+]s - concentration of ion A+ at the surface
X- - counterions
E+ - electrolyte ions
Q - concentration of the excess charge
CA - concentration of analyte at the surface
CE - concentration of electrolyte at the surface
i - bulk concentration
Several authors [17,18] have looked at the application of this equation to the effects of mobile phases but it has not been applied to the ion suppression associated with the competitive ions generated from the stationary phase.
Initial studies associated with the coupling of LC to MS suggested that there was little or no requirement for a separation prior to the detector. However, it was soon realised that coelution of components into the detector would result in a distortion of the process by which ions are formed [19,20,21].Since this observation, the phenomenon of ion suppression or enhancement, which is the reduction or increase of signal response from the MS, has become a major concern to analysts. Significant efforts are employed to reduce the number of components that coelute into the MS by pretreating the samples or employing a separation step. This is particularly the case with complex biological and environmental samples where a considerable number of matrix components are present.
Even with extraction and preconcentration procedures, a target analyte extracted from a complex matrix can still be affected by the remaining components of the matrix [22], as well as other sources. This will lead to inaccurate quantification. There are examples in the academic literature that demonstrate matrix-related effects such as with the analysis of morphine [22], and during the analysis of caffeine where a loss of background ESI signal was observed during repeated injections of a pretreated plasma sample [23]. Researchers have been developing protocols evaluating different sample clean-up methods to minimise matrix effects efficiently. This is achieved by taking account of the matrix types and focusing on the removal of the matrix component [24]. A few researchers have also looked at the contribution of other, method-related components to the levels of ion suppression, with the focus being on the contribution of vials, septa and the extraction consumables to the change in the signal intensity [25].
The use of electrospray has radically changed analytical chemistry, chemistry; however, this increase in analytical power has to be respected and understood to be used effectively. It is the responsibility of the separation scientist to ensure that due diligence is applied to the development of the assay, and that the data produced is truly reflective of what is in the original sample. This starts with the approach to sampling, and sample storage, and continues with the choice of an appropriate sample preparation and separation process to ensure that the detector is giving a response that matches the reality.
References
1. J. B. Fenn, ‘Ion Formation from Charged Droplets: Roles of Geometry, Energy, and Time,’ Journal of the American Society for Mass Spectrometry, vol. 4, no. 7, pp. 524-535, 1993.
2. B. X. Huang, H-Y Kim and C. Dass, ‘Probing three-dimensional structure of bovine serum albumin by chemical cross-linking and mass spectrometry,’ Journal of the American Society for Mass Spectrometry, vol. 15, no. 8, pp. 1237-1247, 2004.
3. Hua Xu, Liwen Zhang and Michael A. Freitas, ‘Identification and Characterization of Disulfide Bonds in Proteins and Peptides from Tandem MS Data by Use of the MassMatrix MS/MS Search Engine,’ Journal of Proteome Research, vol. 7, no. 01, pp. 138-144, 2008.
4. Walter L. Siqueira, Weimin Zhang, Eva J. Helmerhorst, Steven P. Gygi and Frank G. Oppenheim, ‘Identification of Protein Components in in vivo Human Acquired Enamel Pellicle Using LC−ESI−MS/MS’.
5. Atsushi Yamamoto, Naoya Kakutani, Kohji Yamamoto, Toshikazu Kamiura and Hidekazu Miyakoda, ‘Steroid Hormone Profiles of Urban and Tidal Rivers Using LC/MS/MS Equipped with Electrospray Ionization and Atmospheric Pressure Photoionization Sources,’ Environmental Science & Technology, vol. 40, no. 13, pp. 4132-4137, 2006.
6. Y. Chen, F. Al-Taher, R. Juskelis, J. W. Wong, K. Zhang, D. G. Hayward, J. Zweigenbaum, J. Stevens and J. Cappozzo, ‘Multiresidue Pesticide Analysis of Dried Botanical Dietary Supplements Using an Automated Dispersive SPE Cleanup for QuEChERS and High-Performance Liquid Chromatography–Tandem Mass Spectrometry,’ Journal of Agricultural and Food Chemistry, vol. 60, no. 40, p. 9991–9999, 2012.
7. Meritxell Gros, Mira Petrović, Damiá Barceló, ‘Development of a multi-residue analytical methodology based on liquid chromatography–tandem mass spectrometry (LC–MS/MS) for screening and trace level determination of pharmaceuticals in surface and wastewaters,’ Talanta, vol. 70, no. 4, pp. 678-6690, 2006.
8. J.-S. S. J.-K. M. a. J.-H. K. J.-H. Kim, ‘Multi-residue Method Development of 8 Benzoylurea Insecticides in Mandarin and Apple Using High Performance Liquid Chromatography and Liquid Chromatography-Tandem Mass Spectrometry,’ Journal of the Korean Society for Applied Biological Chemistry, vol. 56, no. 1, pp. 47-54, 2013.
9. J. Wang, T. Zhang, Z. Gong, Y. Gao, J. Wang and Y. Zhang, ‘Determination of Eight Benzoylurea Insecticides in High-Fat Foodstuff Samples by Gel Permeation Chromatography Followed by High-Performance Liquid Chromatography-Tandem Mass Spectrometry,’ Food Analytical Methods, vol. 10, no. 9, pp. 3098-3105, 2017.
10. I. R. Pizzutti, A. d. Kok, R. Zanella, M. B.Adaime, M. Hiemstra, C. Wickert and O. D. Prestes, ‘Method validation for the analysis of 169 pesticides in soya grain, without clean up, by liquid chromatography–tandem mass spectrometry using positive and negative electrospray ionization,’ Journal of Chromatography A, vol. 1142, no. 2, pp. 123-136, 2007.
11. M. Dole, L. Mach, R. L. Hines, R. C. Mobley, L. D. Ferguson, M. B. Alice, L.L. Mack, L. L. Mack , ‘Molecular beams of macroions,’ The Journal of Chemical Physics, vol. 49, no. 5, pp. 2240-2249 , 1968.
12. T. B. Iribane JV, ‘On the evaporation of small ions from charged droplets,’ Journal of Chemical Physics, vol. 64, no. 6, pp. 2287-2294, 1976.
13. Xuanfeng Yue, Siavash Vahidi, Lars Konermann, ‘Insights into the Mechanism of Protein Electrospray Ionization From Salt Adduction Measurements,’ Journal of The American Society for Mass Spectrometry, vol. 25, no. 8, pp. 1322-1331, 2014.
14. Anthony T. Iavarone and Evan R. Williams, ‘Mechanism of Charging and Supercharging Molecules in Electrospray Ionization,’ Journal of the American Chemical Society, vol. 125, no. 8, pp. 2319-2327, 2003.
15. Micah T. Donor, Simon A. Ewing, Muhammad A. Zenaidee, William A. Donald, James S. Prell, ‘Extended Protein Ions Are Formed by the Chain Ejection Model in Chemical Supercharging Electrospray Ionization,’ Analytical Chemistry, vol. 89, no. 9, pp. 5107-5114, 2017.
16. L. Tang and P. Kebarle, ‘Effect of the conductivity of the electrosprayed solution on the electrospray current. Factors determining analyte sensitivity in electrospray mass spectrometry,’ Anal. Chem., vol. 64, pp. 2709-2715, 1991.
17. T. L. Constantopoulos, G. S. Jackson and C. G. Enke, ‘Effects of salt concentration on analyte response using electrospray ionization mass spectrometry,’ J. Am. Soc. Mass Spectrom., vol. 10, pp. 625-634, 1999.
18. L. Du and R. L. White, ‘Improved partition equilibrium model for predicting analyte response in electrospray ionisation mass spectrometry,’ J. Mass Spectrom, vol. 44, pp. 222-229, 2009.
19. D. Remane, M. R. Meyer, D. K. Wissenbach and H. H. Maurer, ‘Ion suppression and enhancement effects of co-eluting analytes in multi-analyte approaches: systematic investigation using ultra-high-performance liquid chromatography/mass spectrometry with atmospheric-pressure chemical ionization or electrospray ionizat,’ Rapid Communications in Mass Spectrometry, vol. 24, no. 21, pp. 3103-3108, 2010.
20. Lars Konermann, Elias Ahadi, Antony D. Rodriguez, Siavash Vahidi, ‘Unraveling the Mechanism of Electrospray Ionization,’ Analytical Chemistry, vol. 85, no. 1, pp. 2-9, 2013.
21. G. Shi, ‘Application of co-eluting structural analog internal standards for expanded linear dynamic range in liquid chromatography/electrospray mass spectrometry,’ Rapid Communications in Mass Spectrometry, vol. 17, no. 3, pp. 202-206, 2003.
22. F. Michopoulos, A.M. Edge, Y-T Hui, T. Liddicoat, G. Theodoridis and I.D. Wilson, ‘Extraction methods for the removal of phospholipids and other endogenous material from a biological fluid,’ Bioanalysis, vol. 3, no. 24, pp. 2747-2755, 2011.
23. P. J. Larger, M. Breda, D. Fraier, H. Hughes and C. A. James, ‘Ion-suppression effects in liquid chromatography–tandem mass spectrometry due to a formulation agent, a case study in drug discovery bioanalysis,’ Journal of Pharmaceutical and Biomedical Analysis, vol. 39, no. 1-2, pp. 206-216, 2005.
24. R. King, R. Bonfiglio, C. Fernandez-Metzler, Cynthia Miller-Stein and Timothy Olah, ‘Mechanistic investigation of ionization suppression in electrospray ionization,’ Journal of the American Society for Mass Spectrometry, vol. 11, no. 11, pp. 942-950, 2000.
25. H. Mei, N. C, X. X, W. S, N. K and K. WA, ‘Investigation of matrix effects in bioanalytical high-performance liquid chromatography/tandem mass spectrometric assays: application to drug discovery,’ Rapid Communications in Mass Spectrometry, vol. 97, no. 1, pp. 97-103, 2002.




















-(1).jpg)