Ion chromatography (IC)
Published over 7 years ago. See the latest and most current information on Ion chromatography (IC).
Direct Analysis in Real Time (DART) mass spectrometry was originally introduced [1] as an ambient ionisation method [2] requiring little or no sample preparation. Ionisation with helium DART gas produces primarily protonated or deprotonated molecules directly from solid, liquids or gases. This is still the most common way to use the DART source, but in the past 16 years, new modes of operation and sample handling have evolved that greatly increase the versatility of the technique. In this article, we describe how sample handling methods such in-situ derivatisation, solid-phase microextraction, and thermal desorption and pyrolysis are applied to increase the range of sample types that can be analysed by DART. New modes of operation such as dopant-assisted argon DART and oxygen adduct formation make it possible to detect nonpolar samples and to detect compounds that would not normally be ionised by helium DART. Lastly, the combination of DART with high-pressure liquid chromatography may offer a solution to the problem of ion suppression by nonvolatile buffers.
DART Principle
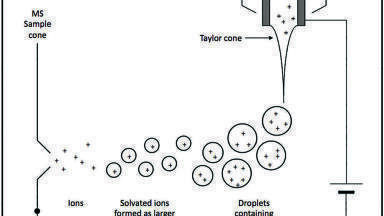
The DART ion source uses a glow discharge in a gas stream to generate long-lived excited-state neutral atoms or molecules (aka ‘metastables’). Penning ionisation [3] produces a positive ion and an electron when the excited-state atoms in the gas stream interact with a sample or substrate with a lower ionisation energy than the internal energy of the metastable atoms M*. The first substrate encountered by the DART gas is often atmospheric moisture or oxygen (Figure 1). Positive-ion mode with helium DART gas typically results in the formation of protonated water clusters, which can transfer a proton to any samples S with a higher proton affinity than water. In negative-ion mode, oxygen undergoes electron capture with the Penning electrons. Oxygen anions can remove a proton from acidic compounds, or in some cases, attach to form oxygen anion adducts.
The DART gas is generally heated to desorb low-volatility samples. Although water and oxygen ions are the most common reagent ions, dopants can be added to the DART gas stream to modify the ionisation chemistry. Matrix effects (suppression or enhancement) can occur if interferences in the sample influence ionisation of the target compound.
All of the examples shown in this article were obtained with a JEOL AccuTOF-DART mass spectrometer, which permits the DART source to be positioned within approximately a centimetre of the mass spectrometer atmospheric pressure interface sampling (API) orifice without additional interface hardware. This is required for the oxygen adduct formation experiment, which cannot be carried out without direct access to the vacuum orifice of the API.
Sample Preparation
While many samples can be analysed directly by DART, rapid and inexpensive sample preparation methods greatly enhance the ability to detect trace components, minimise suppression effects, and permit detection of thermally labile compounds.
In-situ derivatisation
Hot excited-state atoms in the DART gas stream accelerate derivatising reactions, so derivatisation can often be carried out rapidly in the DART gas stream. This is useful for samples that tend to decompose under DART conditions, such as glycosylated or phosphorylated compounds. The most common in-situ derivatisation reactions in DART are thermal hydrolysis and methylation (THAM) with tetramethylammonium hydroxide [4-7] and silylation with common silylating reagents such as BSTFA (N,O-Bis(trimethylsilyl)trifluoroacetamide) and MSTFA (N-Methyl-N-trimethylsilyltrifluoroacetamide) with trichloromethylsilane (TMCS) catalyst. These reagents are typically added onto the sealed tip of a disposable melting point tube together with the sample. The sample and reagent are then suspended in the DART gas stream for analysis.
Oligosaccharides such as cyclodextrins undergo thermal decomposition by DART and cannot be analysed directly without derivatisation. Figure 2a shows a mass spectrum of γ-cyclodextrin (C48H80O40) measured by touching the sealed end of a melting point tube to the powdered sample. Three microlitres of the methylating agent 2.5% tetramethylammonium hydroxide (TMAH) in methanol were applied onto the tip of the melting point tube with the sample. The sample and methylating agent were suspended in the DART gas stream (helium with a gas heater setting of 350°C), and complete methylation occurred within seconds. The permethylated cyclodextrin was observed as the trimethylammonium adduct [C48H80O40 + 24CH2 + (CH3)3NH]+.
The plant hallucinogen psilocybin (C12H17N2O4P) undergoes dephosphorylation with DART, making it indistinguishable from psilocin (C12H16N2O). By adding a drop of MSTFA (1% TMCS) onto the melting point tube with the sample, silylated psilocybin is readily detected (Figure 2b).
Solid-phase microextraction
Solid-phase microextraction (SPME) is easily combined with DART to screen for drugs and environmental contaminants present at low concentrations. SPME is a convenient way to analyse headspace vapours by DART. An early application of SPME to DART in our lab was to detect volatile esters from ripening bananas [8]. SPME/DART was applied to beer brand profiling using headspace vapour [9] and to the detection of cocaine and methadone [10]. Recently, SPME/DART was used to detect transient and reactive volatiles emitted when the root of the Mimosa pudica plant is disturbed [11]. Other forms of rapid sample cleanup have been reported with DART. Microextraction with a packed sorbent (MEPS) was combined with DART to detect cocaine metabolites in urine [12]. Two independent investigations used stir-bar sorptive extraction (SBSE) to detect part-per-trillion contaminants in water [13-15]. Disposable pipette extraction was also applied to the detection of drugs in urine [16].
For analysing trace compounds, SPME has two advantages for DART analysis: it concentrates the sample and removes suppressing interferences. The classic example of sample suppression in DART is the loss of signal for oxazepam in urine due to the presence of excess creatinine. Stout and Ropero-Miller showed that with sufficiently high concentrations of creatinine, oxazepam could not be reliably detected at levels of 100 ppm [17]. To test the application of SPME to toxicological screening for drugs in urine, 17 drugs (including oxazepam) were spiked into urine at concentrations ranging from 0.3 ppb to 300 ppb. Without SPME, only methoxyamphetamine and codeine could be detected at the 300 ppb level. With SPME all of the drugs are detected at 300 ppb and 30 ppb, and some are detected at 3 ppb. SPME/DART has also been applied to the trace detection of synthetic cathinones in human saliva [18].
Other modes of DART operation
Argon DART
Helium is the most commonly used DART gas; although other gases can be used (Chapter 2 in reference [2]). Nitrogen can be useful for target compound identification, but it does not heat the sample as effectively as helium and its higher reactivity relative to helium makes it problematic for identification of unknowns [19]. Neon works by the same mechanism as helium, but its higher cost limits its utility. Argon is a particularly interesting DART gas because the internal energy of metastable argon (Ar*) is 11.55 eV. This is not enough energy to ionise water or oxygen, so as an atmospheric pressure ion source, argon DART is energetically equivalent to atmospheric pressure photoionisation (APPI). Therefore, it requires the use of a dopant with an ionisation energy lower than 11.55 eV. Argon DART was first used with a mixed dopant consisting of acetyl acetone and pyridine for the selective ionisation of melamine in powdered milk [20]. Yang and coworkers showed that argon DART with certain dopants (absolute ethyl alcohol, methanol, fluorobenzene, or acetone) showed reduced fragmentation for labile compounds [21]. In our laboratory, we found that argon with chlorobenzene dopant produces molecular ions M+• instead of protonated molecules [M + H]+ for compounds with relatively low ionisation energies [22]. This enables us to selectively detect compounds that might otherwise be suppressed in a complex matrix. An example is shown in Figure 4 for the detection of β-carotene in the hexane extract from a carrot. Protonated β-carotene (m/z 537.446) is not visible in the helium DART mass spectrum of the extract (Figure 4a) because it is either suppressed or masked by diglycerides present in the same mass range.
O2- attachment for nonpolar compounds and alcohols
Normal DART methods are not suitable for the analysis of saturated hydrocarbons and alcohols. Saturated hydrocarbons do not form protonated molecules (although molecular ions can rarely be observed under very dry conditions [23]). Unless other functional groups are present, alcohols tend to lose water upon protonation. One solution is to derivatise the alcohols [24.25] but this is difficult to do for trace alcohols in the presence of other compounds.
An alternative approach is oxygen anion adduct formation with DART. Samples dissolved in a volatile solvent such as hexane are aspirated directly into vacuum through the mass spectrometer sampling orifice while the DART is operated in negative-ion mode as a source of O2-. Because large alkanes are highly polarisable, O2- can attach to the positive end of the induced dipole to form adducts [M + O2]- without fragmentation [26]. Rapid expansion into vacuum cools the weakly bound adducts so that they can be detected. Alcohols exhibit the same behaviour, making it easy to detect them without derivatisation. Oxygen anion adduct formation was used to identify blowfly species from puparial casings based upon differences in their hydrocarbon profiles [27].
Figure 5 shows the oxygen adduct/DART mass spectrum of a hexane/ethanol extract from the inside surface of cardboard packaging for a commercial pancake batter mix. The base peak is octadecanol with
[M + O2]— at m/z 302.281. Palmitic and stearic acids are detected as the deprotonated molecules [M – H]— at m/z 255.232 and 283.263, respectively. Mineral oil is detected as a series of peaks corresponding to the oxygen adducts [CnH2n+2 + O2]— for saturated hydrocarbons with 23 to 40 carbons. The relative abundance for the mineral oil peaks is low because oxygen adduct formation for hydrocarbons is not as sensitive as for alcohols. These assignments were confirmed by an independent study in our laboratory with comprehensive gas chromatography and high-resolution time-of-flight mass spectrometry (GCxGC/HRTOFMS).
Thermal Desorption and Pyrolysis with DART
By using high gas temperatures (>350°C), DART can be operated in a pyrolysis mode to characterise large molecules such as industrial polymers that cannot be ionised directly by DART. A better approach is to use a thermal desorption and pyrolysis attachment (‘ionRocket’ from Biochromato LLC, Fujisawa, Kanagawa-ken, Japan) for controlled heating of samples. Samples are deposited onto disposable copper sample stubs, mounted on a heating element, and heated gradually, typically from ambient to 600°C at a rate of 100°C min-1. This results in thermal separation of polymer additives followed by pyrolysis of the base polymer. Thermal desorption and pyrolysis DART (TDP/DART) has been used to characterise polylactic acid in 3D-printer feedstock [28], for forensic identification of automotive paint chips [29], forensic identification of sexual lubricants [30] and ignitable liquids in complex matrices [31]. The TDP accessory also facilitates the analysis of fibres and powders that are otherwise difficult to contain in the DART gas stream.
Figure 6 shows the reconstructed ion current chromatograms corresponding to thermal desorption profiles for different components in a 0.5 mm-diameter particle of duct tape and their corresponding DART mass spectra. At temperatures less than about 200°C, the plasticiser diethyl phthalate is observed. Between 200°C and 400°C, we observe a complex pattern of peaks including a peak with an elemental composition consistent with abietic acid. Around 400°C, we observe pyrolysis fragments from polyisoprene (the rubber-based adhesive) and at the highest temperature, we detect the pyrolysis fragments from the low-density polyethylene backing. Peaks associated with pyrolysis of the cotton mesh fibres appear between 500°C and 540°C (not shown).
Chromatography with DART
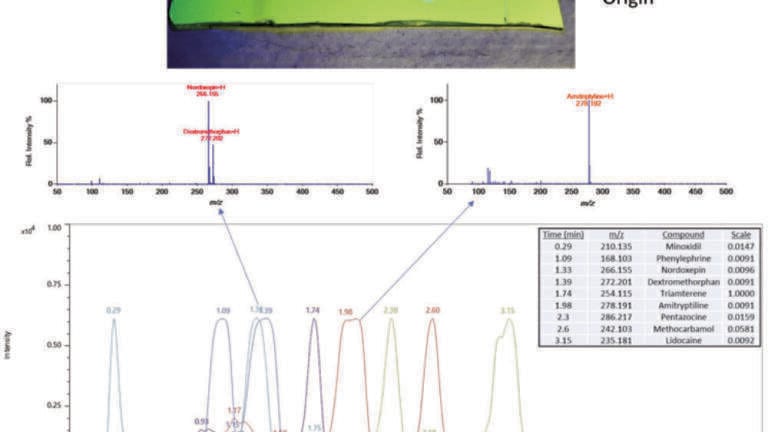
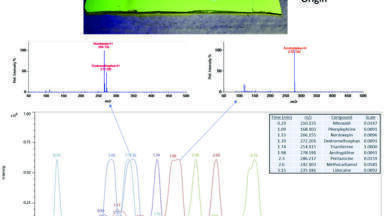
In the absence of chromatography, DART analysis relies on the specificity of the mass spectrometer - that is, resolving power, mass accuracy and tandem mass spectrometry (MS/MS) - to achieve specificity. However, DART has been combined with the most common forms of chromatography including thin-layer chromatography [32-47], gas chromatography [23], liquid chromatography [48,49] and capillary electrophoresis [50]. Of these, the coupling with thin-layer chromatography has been the most common. Figure 7 shows the extracted ion chromatograms for 9 drugs separated on a silica TLC plate with ethyl acetate/methanol/ammonium hydroxide (85:10:5). The TLC plate was cut to a 1 cm width prior to analysis. After separation, the plate was sprayed with 4% glycerol in methanol to facilitate desorption of the drugs from the silica substrate and then scanned horizontally through the DART gas stream at a rate of 3 mm s-1 using the DART-SVP linear rail (IonSense LLC, Saugus MA USA). The DART gas heater was set to 450°C. The chromatograms are normalised to show all compounds on the same scale because the response of the weakest signal is only 1/100 relative to the strongest signals. In contrast to fluorescence (inset), DART analysis reveals all 9 drugs.
The two publications from Johannes Kepler University [48,49] demonstrated the surprising observation that HPLC/DART does not exhibit sample suppression from nonvolatile buffers (such as phosphates), a problem that plagues common HPLC/MS methods such as APCI and ESI. The lack of a commercially available HPLC/DART interface may explain why HPLC/DART has not been more widely used. Given the simplicity of the interface: a zero dead volume connection, stainless steel capillary, mounting brackets and a solvent drain, it is not clear why a commercial interface has not been offered. The problem may be circular: the development may be waiting for customer demand, yet customer demand will not occur until an interface is available. Perhaps this situation will change in the near future and DART will find a place among LC/MS detection methods.
Conclusion
Direct Analysis in Real Time (DART) mass spectrometry can be used for more than simply exposing a sample to the heated DART gas stream. When combined with simple sample handling and sample preparation methods such as in-situ derivatisation, solid-phase microextraction, and thermal desorption and pyrolysis, DART selectivity and detection limits can be greatly improved. Dopant-assisted argon DART and oxygen adduct formation are two variations on DART that permit the selective detection of nonpolar compounds such as saturated hydrocarbons. While DART is often used as a stand-alone ion source, it has been combined with the most common forms of chromatography with characteristics that complement other mass spectrometric detection methods.
References
1. Cody, R. B.; Laramee, J. A.; Durst, H. D. Versatile New Ion Source for the Analysis of Materials in Open Air under Ambient Conditions; Analytical Chemistry 2005, 77, 2297-2302.
2. Domin, M. A.; Cody , R. B. Ambient Ionization Mass Spectrometry; Royal Society of Chemistry, 2015.
3. Penning, F. M. Über Ionisation durch Metastabile Atome; Naturwissenschaften 1927, 15, 818.
4. Pierce, C. Y.; Barr, J. R.; Cody, R. B.; Massung, R. F.; Woolfitt, A. R.; Moura, H.; Thompson, H. A.; Fernandez, F. M. Ambient generation of fatty acid methyl ester ions from bacterial whole cells by direct analysis in real time (DART) mass spectrometry; Chem. Commun. 2007, 807 - 809.
5. Fraser, D.; DeRoo, C. S.; Cody, R. B.; Armitage, R. A. Characterization of blood in an encrustation on an African mask: spectroscopic and direct analysis in real time mass spectrometric identification of haem; Analyst 2013, 138, 4470-4474.
6. Kim, H. J.; Park, S. R.; Jang, Y. P. Extraction-free In situ Derivatisation of Timosaponin AIII Using Direct Analysis in Real Time TOF/MS; Phytochemical Analysis 2014, 25, 373-377.
7. Zeng, S.; Wang, L.; Chen, T.; Qu, H. On-line coupling of macroporous resin column chromatography with direct analysis in real time mass spectrometry utilizing a surface flowing mode sample holder; Analytica Chimica Acta 2014, 811, 43-50.
8. JEOL USA Inc. Using Solid Phase Microextraction with AccuTOF-DART® for Fragrance Analysis (Application Note).
9. Cajka, T.; Riddellova, K.; Tomaniova, M.; Hajslova, J. Recognition of beer brand based on multivariate analysis of volatile fingerprint; Journal of Chromatography A 2010, 1217, 4195-4203.
10. Rodriguez-Lafuente, A.; Mirnaghi, F.; Pawliszyn, J. Determination of cocaine and methadone in urine samples by thin-film solid-phase microextraction and direct analysis in real time (DART) coupled with tandem mass spectrometry; Analytical and Bioanalytical Chemistry 2013, 1-5.
11. Musah, R. A.; Lesiak, A. D.; Maron, M. J.; Cody, R. B.; Edwards, D.; Fowble, K. L.; Dane, A. J.; Long, M. C. Mechanosensitivity Below Ground: Touch-Sensitive Smell-Producing Roots in the “Shy Plant,” Mimosa pudica L; Plant Physiology 2015, 170, 1075-1089.
12. Jagerdeo, E.; Abdel-Rehim, M. Screening of Cocaine and Its Metabolites in Human Urine Samples by Direct Analysis in Real-Time Source Coupled to Time-of-Flight Mass Spectrometry After Online Preconcentration Utilizing Microextraction by Packed Sorbent; Journal of the American Society of Mass Spectrometry 2009, 20, 891-899.
13. Loftin, K. B. DEVELOPMENT OF NOVEL DART™ TOFMS ANALYTICAL TECHNIQUES FOR THE IDENTIFICATION OF ORGANIC CONTAMINATION ON SPACEFLIGHT-RELATED SUBSTRATES AND AQUEOUS MEDIA. Ph.D. Thesis, University of Central Florida2009.
14. Haunschmidt, M.; Klampfl, C. W.; Buchberger, W.; Hertsens, R. Rapid identification of stabilisers in polypropylene using time-of-flight mass spectrometry and DART as ion source; Analyst 2010, 135, 80-85.
15. Haunschmidt, M.; Klampfl, C.; Buchberger, W.; Hertsens, R. Determination of organic UV filters in water by stir bar sorptive extraction and direct analysis in real-time mass spectrometry; Analytical and Bioanalytical Chemistry 2010, 397, 269-275.
16. Cody , R. B.; Dane, A. J.; Brewer, W. E. In 59th Annual Conference on Mass Spectrometry and Allied Topics: Denver, CO, 2011.
17. Stout , P. R.; Ropero-Miller, J. D. In NCJRS Abstract, 2008.
18. Tully, K.; Musselman, B.; Morrison, J. In 250th ACS National Meeting: Boston, MA, 2015.
19. Song, L.; Chuah, W. C.; Lu, X.; Remsen, E.; Bartmess, J. E. Ionization Mechanism of Positive-Ion Nitrogen Direct Analysis in Real Time; Journal of the American Society for Mass Spectrometry 2018, 29, 640-650.
20. Dane, A. J.; Cody, R. B. Selective Ionization of Melamine in Powdered Milk by Using Argon Direct Analysis in Real Time (DART) Mass Spectrometry; The Analyst 2009, 135, 696-699.
21. Yang, H.; Wan, D.; Song, F.; Liu, Z.; Liu, S. Argon Direct Analysis in Real Time Mass Spectrometry in Conjunction with Makeup Solvents: A Method for Analysis of Labile Compounds; Analytical Chemistry 2013, 85, 1305-1309.
22. Cody, R. B.; Dane, A. J. Dopant-assisted direct analysis in real time mass spectrometry with argon gas; Rapid Communications in Mass Spectrometry 2016, 30, 1181-1189.
23. Cody, R. B. The Observation of Molecular Ions and Analysis of Nonpolar Compounds with the Direct Analysis in Real Time Ion Source; Analytical Chemistry 2009, 81, 1101-1107.
24. Laramée, J. A.; Durst, H. D. N., J. Michael; Connell, T. R. Alcohols Can Now Be Analyzed by a Direct Analysis in Real-Time Method: Applications for Chemical Warfare Agent Synthesis; American Laboratory 2009, 41, 24-27.
25. Laramee, J. A.; Durst, H. D.; Nilles, J. M.; Connell, T. R. An Improved Protocol for the Analysis of Alcohols by Direct Analysis in Real Time Mass Spectrometry; American Laboratory 2009, 41, 25-27.
26. Cody, R. B.; Dane, A. J. Soft Ionization of Saturated Hydrocarbons, Alcohols and Nonpolar Compounds by Negative-Ion Direct Analysis in Real-Time Mass Spectrometry; Journal of The American Society for Mass Spectrometry 2013, 24, 329-334.
27. Musah, R. A.; Espinoza, E. O.; Cody, R. B.; Lesiak, A. D.; Christensen, E. D.; Moore, H. E.; Maleknia, S.; Drijfhout, F. P. A High Throughput Ambient Mass Spectrometric Approach to Species Identification and Classification from Chemical Fingerprint Signatures; Sci. Rep. 2015, 5.
28. Cody, R. B.; Fouquet, T. “Reverse Kendrick Mass Defect Analysis”: Rotating Mass Defect Graphs to Determine Oligomer Compositions for Homopolymers; Analytical Chemistry 2018.
29. Marić, M.; Marano, J.; Cody, R. B.; Bridge, C. DART-MS: A New Analytical Technique for Forensic Paint Analysis; Analytical Chemistry 2018.
30. Bridge, C.; Marić, M. Temperature-Dependent DART-MS Analysis of Sexual Lubricants to Increase Accurate Associations; Journal of the American Society for Mass Spectrometry 2019.
31. Barnett, I.; Bailey, F. C.; Zhang, M. Detection and Classification of Ignitable Liquid Residues in the Presence of Matrix Interferences by Using Direct Analysis in Real Time Mass Spectrometry; Journal of Forensic Sciences, 0.
32. Morlock, G.; Schwack, W. Determination of isopropylthioxanthone (ITX) in milk, yoghurt and fat by HPTLC-FLD, HPTLC-ESI/MS and HPTLC-DART/MS; Analytical and Bioanalytical Chemistry 2006, 385, 586-595.
33. Morlock, G.; Ueda, Y. New coupling of planar chromatography with direct analysis in real time mass spectrometry Journal of Chromatography A 2007, 1143, 243-251
34. Morlock, G.; Ueda, Y. Coupling Planar Chromatography with Time-of-Flight Mass Spectrometry Using an Open-Air Ion Source; LCGC: The Peak 2007, 7-13.
35. Dytkiewitz, E.; Morlock, G. E. Analytical Strategy for Rapid Identification and Quantification of Lubricant Additives in Mineral Oil by High-Performance Thin-Layer Chromatography with UV Absorption and Fluorescence Detection Combined with Mass Spectrometry and Infrared Spectroscopy; Journal of AOAC INTERNATIONAL 2008, 91, 1237-1244.
36. Alpmann, A.; Morlock, G. Rapid and sensitive determination of acrylamide in drinking water by planar chromatography and fluorescence detection after derivatization with dansulfinic acid; Journal of Separation Science 2008, 31, 71-77.
37. Morlock, G.; Schwack, W. Planar Chromatography - Back to the future.; LC/GC Europe 2008, 366-371.
38. Chernetsova, E.; Morlock, G. Ambient desorption ionization mass spectrometry (DART, DESI) and its bioanalytical applications; Bioanalytical Reviews 2011, 3, 1-9.
39. Chernetsova, E. S.; Revelsky, A. I.; Morlock, G. E. Some new features of Direct Analysis in Real Time mass spectrometry utilizing the desorption at an angle option; Rapid Communications in Mass Spectrometry 2011, 25, 2275-2282.
40. Chernetsova, E. S.; Morlock, G. E. Ambient desorption ionization mass spectrometry (DART, DESI) and its bioanalytical applications; Bioanalytical Reviews 2011, 3, 1-9.
41. Chernetsova, E. S.; Morlock, G. E.; Revelsky, I. A. DART mass spectrometry and its applications in chemical analysis; Russian Chemical Review 2011, 80, 235.
42. Chernetsova, E. S.; Morlock, G. E. Determination of drugs and drug-like compounds in different samples with direct analysis in real time mass spectrometry; Mass Spectrometry Reviews 2011, 30, 875-883.
43. Chernetsova, E. S.; Morlock, G. E. Assessing the capabilities of direct analysis in real time mass spectrometry for 5-hydroxymethylfurfural quantitation in honey; International Journal of Mass Spectrometry 2012, 314, 22-32.
44. Chernetsova, E. S.; Bromirski, M.; Scheibner, O.; Morlock, G. E. DART-Orbitrap: a novel mass spectrometric approach for the identification of phenolic compounds in propolis; Analytical and Bioanalytical Chemistry 2012, 403, 2859-2867.
45. Morlock, G. E.; Chernetsova, E. S. Coupling of planar chromatography with direct analysis in real time mass spectrometry (DART-MS) cent.eur.j.chem. 2012, 10, 703-710.
46. Smith, N. J.; Domin, M. A.; Scott, L. T. HRMS Directly From TLC Slides. A Powerful Tool for Rapid Analysis of Organic Mixtures; Org. Lett. 2008, 10, 3493-3496.
47. Howlett, S. E.; Steiner, R. R. Validation of Thin Layer Chromatography with AccuTOF-DART™ Detection for Forensic Drug Analysis*; Journal of Forensic Sciences 2011, 56, 1261-1267.
48. Eberherr, W.; Buchberger, W.; Hertsens, R.; Klampfl, C. W. Investigations on the Coupling of High-Performance Liquid Chromatography to Direct Analysis in Real Time Mass Spectrometry; Analytical Chemistry 2010, 82, 5792-5796.
49. Beißmann, S.; Buchberger, W.; Hertsens, R.; Klampfl, C. W. High-performance liquid chromatography coupled to direct analysis in real time mass spectrometry: Investigations on gradient elution and influence of complex matrices on signal intensities; Journal of Chromatography A 2011, 1213, 7.
50. Chang, C.; Xu, G.; Bai, Y.; Zhang, C.; Li, X.; Li, M.; Liu, Y.; Liu, H. Online Coupling of Capillary Electrophoresis with Direct Analysis in Real Time Mass Spectrometry; Analytical Chemistry 2013, 85, 170-176.





















-(1).jpg)