HPLC, UHPLC
Published over 5 years ago. See the latest and most current information on HPLC, UHPLC.
Acrylamide was first discovered in foodstuffs in 2002 [1,2], causing widespread alarm due to its classification as a probable carcinogen [3]. By 2017, regulatory authorities established benchmark levels and recommended that companies continue to monitor and minimise the acrylamide levels in heat processed foods. There has since been an increased demand for a reliable and accurate method for acrylamide detection. Traditional methods using liquid chromatography with tandem mass spectrometry (LC-MS/MS) have faced numerous challenges due to the highly polar nature of acrylamide, the need for extensive sample clean-up and the general lack of reliability in the results. An improved method for acrylamide detection using a novel two-step extraction procedure is proposed in this article. With better selectivity, excellent recovery and a higher signal/noise ratio, this method can be applied to monitor acrylamide levels in a range of foodstuffs.
Traces of acrylamide discovered in foodstuffs in 2002 [1,2] caused immediate concern due to potential health risks. Even before its discovery in foodstuffs, the toxicological effects of acrylamide had already been reported. Acrylamide is considered a neurotoxicant, reproductive toxin and mutagen [3]. The Maillard reaction between amino acids and reducing sugars in carbohydrate-rich and heat-processed foods [4] is responsible for generating higher concentrations of acrylamide in these foods.
After extensive exposure assessments and screening between 2007 and 2011, the US Food and Drug Administration (FDA) [5] and European Food Safety Authority [6] set indicative values for acrylamide in a variety of food products and recommended companies to monitor the levels of acrylamide as well as adopt approaches to reduce its levels. In 2017, benchmark levels were established for foodstuffs, such as french fries, potato crisps, coffee, baby foods and biscuits, among others [7]. Continued investigations may pose tighter regulations in the future to safeguard consumer health.
Developing a robust analytical method that can accurately and reliably detect and quantify acrylamide levels is important to the food industry to ensure testing meets the required regulations and foods are safe for public consumption.
The nature of acrylamide as an analyte poses numerous challenges in its measurement. Acrylamide is highly polar hence difficult to retain using reverse phase chromatography techniques, and as it lacks a chromophore, conventional UV detection methods are not possible. Adding chemical derivatisation steps can enable UV detection or increase the lipophilicity to make reverse phase chromatography applicable, but makes protocols time-consuming. During MS separation, other small polar compounds of the same mass may cause specificity issues as well as ion suppression.
In addition to this, improper sample preparation steps or failure to clean the column after each injection can result in column fouling, significantly decreasing column lifetime and affecting peak shape. For instance, improper sample clean up for a fatty matrix like potato chips may lead to lipids making their way into the final samples.
An improved assay for accurate quantitation of acrylamide from three different food matrices, namely, potato chips, coffee and baby food is proposed in this article. A novel two-step extraction process is employed. The first step was designed to remove fats and lipophilic compounds by using dichloromethane (DCM) to solubilise and partition them. The second step, involving Supported Liquid Extraction (SLE), refines and enables concentration of the extracts. After developing this method, it was used to determine acrylamide levels in burnt bread.
Sample preparation and two-step extraction
1 g ± 0.05 g of ground potato chips, ground coffee or homogenised baby food were weighed out into 15 mL Thermo Scientific Nunc tubes. The food matrices were fortified with acrylamide calibration standards (10 μL) and the stable labelled acrylamide-d3 internal standard (10 μL). Solvents used to dissolve the standard were allowed to evaporate for 30 minutes, leaving behind the acrylamide residues. 10 mL of water was then added and the tubes were placed on a horizontal flatbed shaker for 30 minutes. 2 mL of dichloromethane was then added, placing the tubes back on the shaker for 10 minutes, followed by centrifugation at 3500 × g for 15 minutes.
A 200 μL aliquot of the top aqueous phase was then added to the appropriate wells of a SLE 96 well plate (200 mg (pH 9)/2 mL) (HyperSep SLE, Thermo Scientific) and allowed to absorb for 15 minutes. When gravity was insufficient, a small pulse of vacuum was sometimes required to draw the sample into the plate. The compounds were eluted with 2 × 750 μL of ethyl acetate/tetrahydrofuran (50/50, v/v) into a 2 mL 96-well plate containing 20 μL of ethylene glycol. A pulse of vacuum was applied to fully dry the plate and it was left to evaporate under N2 at 40°C for approximately 1 hour. Finally, an additional 200 μL of water was added to each well, capped and vortexed, followed by centrifugation at 3500 × g for 15 minutes.
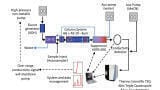
An ultra-high performance liquid chromatography (UHPLC) system (Thermo Scientific™ Vanquish Horizon™) was used for separation with the following solvents: water/formic acid (100/0.5, v/v) as mobile phase A and methanol as mobile phase B. Flow rate was maintained at 0.5 mL/min with a linear gradient curve showing instant change as listed in Table 1. Total run time was 4.5 minutes. The HPLC column (Thermo Scientific™ Hypercarb™ HPLC column (100 × 2.1 mm, 5 μm)) was used due to its novel selectivity and ability to retain highly polar species. It was maintained at 60°C with active pre-heating and still air mode.
For MS/MS, a Triple-Stage Quadrupole Mass Spectrometer (Thermo Scientific™ TSQ Endura™) was used with the parameters listed in Table 2. The m/z values for setting selected reaction monitoring (SRM) transition of the analytes of interest, acrylamide and internal standard, acrylamide-d3, are shown in Table 2. Data was acquired and analysed using a chromatography data system software (Thermo Scientific™ Chromeleon™ Chromatography Data System (CDS) software version 7.2.10).
The calibration model was assessed to be a linear regression with 1/x2 weighting (Figure 1). Eight calibration standards over the range 100–5000 ng/g were freshly prepared in water/ethylene glycol (90/10, v/v). As acrylamide was expected to be found above the LLOQ in many matrices and determining the concentration of acrylamide in many food types in one run was the aim, a non-extracted calibration line was used. Absolute recovery was calculated intra-day for each matrix.
Measuring accuracy and precision with known samples adds reliability to the concentrations obtained from unknowns. To determine accuracy and precision, six different replicates of quality control (QC) samples at four concentrations for all three food matrices were tested over the calibration range. QCs were assessed as non-extracted samples and as extracts in each of the three food types. For the food QCs, the average concentration of acrylamide was determined from three single blank samples of each food type. The average concentration measured in the three single blank samples was added to the nominal concentration to give a new theoretical QC concentration for each food matrix.
Accuracy and precision data of non-extracted samples as well as the three food samples appear in Table 3.
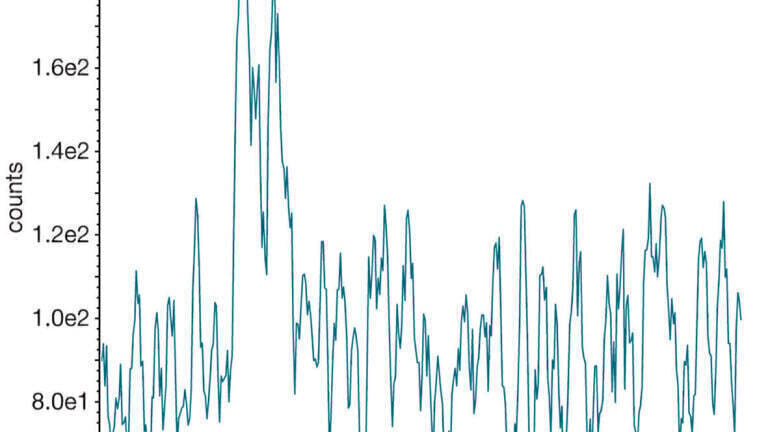
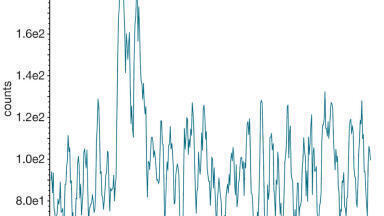
The nominal concentrations were 100 ng/g LLOQ, 250 ng/g low QC, 800 ng/g medium QC, and 4000 ng/g high QC. Intra-batch accuracy and precision was acceptable for all three matrices with bias being <13% and CV <5% at each of the four QC levels in both non-extracted and extracted food QCs, demonstrating that the assay is accurate and precise at the concentrations measured when results are read off a non-extracted calibration line. A representative potato crisp chromatogram at the low QC level is presented in Figure 2.
Figure 2: Example chromatogram at low QC in ground potato crisps.
As measurable concentrations of acrylamide were expected to be seen in each of the sources of food it was only possible to check that the working concentration of internal standard did not contribute to the acrylamide channel due to isotopic impurity. One double blank sample (no acrylamide or internal standard added) prepared in water/ethylene glycol (90/10, v/v) and one single blank sample (internal standard only added) prepared in water/ethylene glycol (90/10, v/v) were chromatographed.
Specificity was proven for acrylamide and acrylamide-d3 as no interfering peaks were detected in the blank samples. A representative chromatogram of a single blank is shown in Figure 3.
Carryover can result from either poor cleaning of the UHPLC system or ‘memory effects’ of analytes that are still adhered to the column from previous injections. The carryover for this system was assessed by injecting two double blank samples after the injection of the highest calibration sample. The area response of the analyte and internal standard in the blank samples was compared to the area responses of the non-extracted LLOQ samples after them. No carryover was present using the Vanquish Horizon UHPLC system as shown in Figure 4.
The evaluation of matrix effects and extraction efficiencies
For complex matrices, such as those encountered in food and beverage analysis, the use of a suitable internal standard is strongly recommended. The internal standard should be added before any manipulation of the samples so it can correct for small volume discrepancies during sample transfers and variations in recovery.
As the internal standard used here was also stable-labelled, it effectively compensated for potential ion suppression or enhancement during MS/MS analysis.
As acrylamide was shown to be present in each of the food types tested, it was not possible to separate the matrix effects and recovery experiments as would normally be the case. Absolute recovery (combined matrix effects and recovery) was calculated by comparing the average peak area for the internal standard in the six low QC replicates, for each of the three sources of food matrix, compared to the internal standard peak area in the non-extracted QCs. This approach was valid given that no peak was visible for the internal standard transition in double blank food matrices. Additionally, as a stable-labelled version of acrylamide was used, it had identical properties throughout the extraction procedure and during UHPLC-MS/MS analysis. Table 4 below represents the absolute recovery from the three foods measured. The combined recovery and suppression, irrespective of the food matrix, was less than 20%.
Once the method for quantitative analysis of acrylamide was tested for reliability using the three food matrices, it was then applied to determine acrylamide concentrations in burnt brown and white toast. As acrylamide is known to form with excessive heating of carbohydrate-rich food, the toasts were burnt to a point where they could still be acceptable for consumption. The whole pieces were then ground into a fine powder. Three replicate samples from each piece underwent the same sample preparation procedure followed by UHPLC-MS/MS analysis.
The results revealed that the concentration of acrylamide in the burnt brown toast was double the levels found in burnt white toast (Table 5). Sample replicates showed minimal variation, suggesting that the prepared samples were homogenous, and the method, highly reproducible.
A simple yet robust two-stage extraction and clean-up procedure significantly improved acrylamide assay quality, making it reproducible across replicates, and resulted in higher recovery and the absence of matrix effects. Table 6 outlines the parameters used in the assay.
Table 6: Summary of the acrylamide assay using the two-step sample extraction process.
Choosing an appropriate column with a higher retention capacity for polar analytes resulted in acceptable retention and better selectivity for acrylamide. The 4.5-minute runtime employing the current parameters offered a significant improvement over previous protocols with runtimes of ~11 minutes, enabling faster testing to support high-throughput projects.
Compared to earlier methods for acrylamide analysis, the two-step extraction coupled with the organic flush prevents column fouling. The higher signal/noise ratio obtained as a result of the improvements in this protocol allows the injection volume to be minimised to 1μL, further reducing the potential for column and system fouling. The updated protocol was suitable to measure acrylamide in a range of foodstuffs.
1. Opinion of the Scientific Committee on Food on new findings regarding the presence of acrylamide in food, European Commission, July 2002 https://ec.europa.eu/food/sites/food/files/safety/docs/sci-com_scf_out131_en.pdf
2. WHO to hold urgent expert consultation on acrylamide in food after findings of Swedish National Food Administration, WHO,
June 2002 https://www.who.int/mediacentre/news/releases/release32/en/
3. Exon JH. A review of the toxicology of acrylamide. J Toxicol Environ Health B Crit Rev. 2006
4. Mottram, D., Wedzicha, B. & Dodson, A. Acrylamide is formed in the Maillard reaction. Nature 2002
5. Guidance for Industry: Acrylamide in Foods, US FDA, 2016 https://www.fda.gov/regulatory-information/search-fda-guidance-documents/guidance-industry-acrylamide-foods
6. Commission Regulation (EU) 2017/2158 of 20 November 2017 establishing mitigation measures and benchmark levels for the reduction of the presence of acrylamide in food. Official Journal of the European Union, 2017 https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX:32017R2158
7. Reducing the presence of acrylamide in food, EUR-Lex, 2018



















-(1).jpg)