HPLC, UHPLC
Published over 5 years ago. See the latest and most current information on HPLC, UHPLC.
Non-targeted screening is a well-established analytical strategy. However, overcoming sample complexity remains a significant challenge. Over the last two decades, high resolution mass spectrometry (HRMS) and liquid chromatography (LC) have evolved, providing increased peak capacity to resolve sample complexity. Ion mobility (IM) separation provides both a third dimension of resolution and collision cross section values (CCSs); an additional analyte descriptor. Here, we review LC-MS technology advances applied to medicinal plant screening. Ultra-performance liquid chromatography ion mobility mass spectrometry (UPLC-IM-MS) reduces analysis cycle time, enhances peak capacity and facilitates identification of isomeric, ‘known’ and ‘known-unknown’ analytes to generate enhanced speciation profiles.
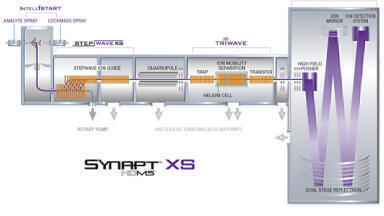
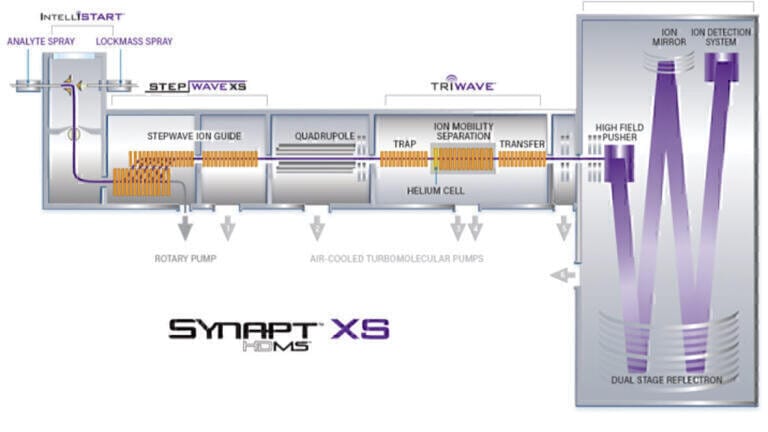
LC-MS techniques which use high resolution mass measurements are important tools for the identification of the constituents in complex samples such as medicinal or agriculturally important plants and generation of a phytochemical profile. Due to technology evolution, over the past two decades there has been an increase in the prevalence of non-targeted screening assays which use accurate mass data. The use of accurate mass measurements for precursor ions, particularly for small molecules, instils greater confidence in compound confirmation, since the elemental composition of a detected analyte can be determined. An increase in identification specificity can also be achieved through generation of accurate mass product ion information. In the case of data-independent non-targeted screening, acquisitions are commonly performed where two data channels are acquired simultaneously (MSE), the first contains precursor ion information, and the second data channel is acquired using a collision energy ramp, to generate fragment ions which can be correlated with their precursor ions on the basis of retention time (tr) alignment. Such a strategy facilitates the generation of accurate mass precursor and fragment ion structural elucidation information, without the requirement to produce a target list. Both informatics and MS instrumentation advances have led to an increase in the use of high-resolution mass spectrometry (HRMS), including orthogonal acceleration time-of-flight (oa-TOF) and quadrupole time of flight (Q-TOF) platforms and are fully described elsewhere [1-4]. A conventional Q-oa-TOF-MS platform incorporates a quadrupole (used for precursor mass selection, MS/MS and quantitation), a collision cell and a time of flight resolving mass analyser, which incorporates reflectron technology to increase the time of flight path length and subsequent mass resolution. For full spectra acquisition the quadrupole is operated in RF-only mode as a wide band-pass filter to transmit a wide mass range. The continuous ion beam is gated and pulsed into an orthogonal accelerator, where ions are accelerated down the flight tube and follow a focused path to the detector. In the mass analyser, lighter ions traverse faster than heavier ions and are resolved based on mass to charge ratio. In a Q-IM-oa-TOF-MS platform an ion mobility separation device is included between the quadrupole and the mass analyser (see Figure 1). Nowadays it is routine to detect thousands of components in a single analysis, which can be followed by retrospective targeted data analysis.
The commercial introduction of sub-2µm particles with systems operating at higher back pressures and high mobile phase linear velocities, have facilitated a reduction in the length of chromatographic gradients, while retaining or increasing peak capacity, and affording improvements in sensitivity [5].
Despite these advances in LC and MS (including MS/MS), it can still be a challenge to confirm the presence of a specific analyte, especially when isomers/structurally related compounds produce the same isomeric product ions and in the presence of co-extracted matrix components. One example is the identification of flavonoids in medicinal plants. Over 4,000 flavonoids have been identified, they are a widespread class of compounds possessing diverse pharmacological and biological properties [6]. The genus Passiflora contains approximately 500 species, each of which contain apigenin/luteolin flavonoid derivatives (C-glycosylflavones), which frequently occur as isomers [7]. By characterising and identifying 8-C and 6-C glycoside critical isomer pair ratios of isoorientin/orientin, isovitexin/vitexin it’s possible to distinguish the four species of Passiflora:- P. edulis, alata, caerulea and incarnata [8]. The two 8-C and 6-C glycoside forms have distinctive isomeric positive ion and negative ion electrospray product ion spectra. However, utilising such specific characteristic analyte information in a data dependent or targeted analysis strategy can be hindered by sample complexity.
Efficacy regulation and assessment of over the counter (OTC) herbal medicines extends beyond phytochemical profiling. Quality control to establish purity and determine the presence of fumigants, pesticides, fungi, mycotoxins and bacteria (salmonella and E. coli) is required to ensure consumer safety. Analysis is required to optimise extraction procedures and the blending of extracts to produce a known quantity of active ingredients within a final product herbal formulation. Environmental conditions can also impact phytochemical make-up. Commodity stability and degradation relating to storage conditions, packaging and humidity need also to be assessed. Efficacy within controlled clinical trials requires standardised extracts and standardised doses. Here, we review advances in LC-MS analysis that can be used to overcome challenges in phytomedicine characterisation and facilitate profiling of phytochemical over-the-counter commodity products.
In this article, we offer insights into how the challenge of sample complexity has been overcome, using the LC-MS technology advances, in particular, the coupling of ultra-performance liquid chromatography with ion mobility mass spectrometry (UPLC-IM-MS), which provides three dimensions of separation; m/z, chromatographic and ion mobility resolution. The commercialisation of IM instrumentation (Waters Corporation, Milford, MA, USA - SYNAPT (2006)) [9,10] has led to a continuous increase in publications citing the use of IM [11,12]. IM is a property of gas phase ions relating to their velocity in a buffer gas relative to an applied electric field, where the IM separation depends on an ion’s mass, charge and shape [13]. Dwivedi et al illustrated a factor of 2 to 10 increase in peak capacity using LC, MS, and IM, depending on the MS and IM resolution [14,15]. An increase in analytical strategies which use collision cross section (CCS) values as an additional descriptor to aid identification have taken place, in combination with MS measurement and tr, they provide a complementary metric [16-18]. Development in chromatographic and mass resolution separation strategy have been employed, to reach an end point where using ion mobility, speciation profiling of Passiflora is extended beyond reliance upon only two known critical isomer pairs (see Figure 2). A ‘known-unknown’ compound is defined as an analyte or feature that is consistently detected and characterised by multi-metric analytical measurements in and across multiple samples [19]. Incorporation of ‘knowns’, ‘known-unknowns’ and identification of ‘known-unknowns’, makes it possible to obtain a comprehensive component marker fingerprint to control the quality of phytomedicines and profile the phytochemical makeup of medicinal plants [20-23].
Sample information:
Preparation of the voucher specimens of all plant materials employed in this study, P. incarnata, P. edulis, P. caerulea and P. alata has previously been described [8]. Extraction was with ethanol/water (2:1 v/v), 1 g plant/10 mL solvent, according to the Brazilian Pharmacopoeia procedure [24].
Chromatographic and Mass Spectrometry Conditions.
High performance liquid chromatography (HPLC)-oa-TOF-MS, UPLC-IM-MS experimental and processing parameters have been described previously elsewhere [8,25-27].
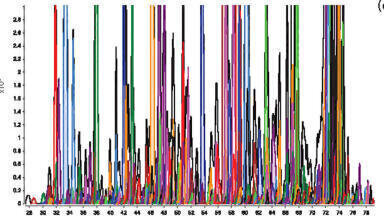
Profiling of Passiflora variants highlights the challenges of complex sample analysis, which are common to numerous areas of research including metabolomics, metabolite identification and food safety (veterinary drug residues, pesticide residues analysis). Incremental advances in mass spectrometry technology and informatics have unleashed the potential of high-specificity, data independent non-targeted screening Three-dimensional resolution comprised of m/z, chromatographic and ion mobility separation is illustrated for Passiflora edulis in Figure 3. UPLC-IM-MS provides an alternate strategy to the 2-D separation techniques discussed by Cao et al and Zhu et al [28,29]. Retention time and drift time (DT) aligned spectra provide specificity akin to that of directed MSMS, however the necessity to perform preselection of target analyte mass using the quadrupole is negated. A CCS value is also generated for all IM resolved precursor analytes, which can enable isomeric species to be distinguished. Observed and expected CCS (Å2) values are routinely reported alongside tr (min) and m/z accurate mass (ppm).
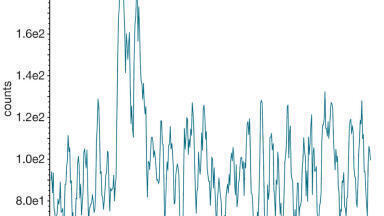
The ability to achieve higher mass resolutions is a major factor in overcoming the challenges of complex sample analysis, since it facilitates the separation of isobaric species which improves m/z specificity. Increases in oa-TOF-MS mass resolution from > 5000 FWHM to > 50000 FWHM are illustrated at m/z 556 [M+H]+ for the TOF-MS system lock mass leucine enkephalin in Figure 4 and compared to unit resolution of a quadrupole MS instrument. The introduction of UPLC facilitated a 66% reduction in total analysis cycle time to 17 mins (50 min using HPLC), for the analysis of Passiflora species (see Figure 4) and chromatographic base peak widths were sharpened from ~ 1min to ~ 0.1 min (for the 17 min UPLC gradient used). The enhanced peak capacity [5] provides further component separation and correspondingly cleaner retention time aligned product ion spectra in MSE experiments. UPLC improves S/N due to the reduction in band broadening, and thus an increase in sensitivity. Extracts analysed using UPLC-IM-MS were diluted 40:1 compared to those used for HPLC-oa-TOF-MS.
Distinctive fragment ion distributions were identified during our initial HPLC-MS investigations using positive mode ESI [8]. At uncharacteristically high collision induced dissociation (CID) collision energies, the isomer pairs formed differentiating isomeric fragment ions. Accurate mass measurement confirmed the elemental measurement and reaffirmed the discovery of characteristic information that could be used to differentiate 6-C and 8-C glycoside isomers, as illustrated in Figure 5 for isoorientin and orientin [8]. Subsequently, using UPLC-IM-MS negative mode ESI, characteristic 6-C and 8-C glycoside fragment ions were identified (see Figure 6) [25]. Comparing Figure 5 and 6, informatics evolution is also illustrated, where detected analyte structure and fragmentation dissociation pathways are incorporated into the targeted data processing results. Despite the improved peak capacity brought on by an evolution of chromatographic and mass resolution, coelution resulting from sample complexity can prevent the acquisition of non-targeted single component fragment ion data. However, ion mobility facilitates an additional dimension of separation. Co-eluting isobaric species, metabolites or isomers (that may produce the same product ions) would produce a mixed fragment ion spectrum (MSE), but when they are additionally separated using ion mobility (HDMSE), single component fragment ion spectra occur.
Investigations into non-targeted authentication profiling using the combined specificity of UPLC-IM-MS and CID have been carried out to provide unambiguous identification of flavonoid analytes TWCCSN2 (CCS values generated using travelling wave ion mobility and a nitrogen buffer gas) values were used to produce unequivocal identification of marker flavonoid isomers (6-C and 8-C-glycosylflavone isomer pairs orientin (187.7 Å2)/isoorientin (198.1 Å2) and vitexin (195.5 Å2)/isovitexin (188.8 Å2)) [25]. Coeluting with structurally related analytes in the complex leaf extracts of Passiflora, the retention times determined for isoorientin and orientin were different by 0.05 min and for isovitexin/vitexin by 0.12 mins. In the Passiflora extracts analysed, the concentrations of isoorientin and orientin are such that they merge to form one chromatographic peak. Using accurate mass measurements alone, these coeluting marker isomers would be indistinguishable, however using both IMS and optimised UPLC conditions, both critical isomer pairs are resolved (see Figure 7) [25]. Not achievable with mass resolution alone, the isomeric product ion spectra shown in Figure 6 are produced from IM resolved chromatographically coeluting orientin and isoorientin isomers shown in Figure 7. Utilising HDMSE, ‘fragment ions’ are generated from a specific retention time/drift time aligned precursor ion, hence are referred to as product ions [26,27]. Detailed profiling of Passiflora species using ion mobility mass spectrometry have previously been described [25].
CCS values can also increase the identification certainty of ‘known-unknowns’ analytes as well as ‘knowns’. UPLC-IM-CID-MS non-targeted specificity, i.e. CCS measurements and distinctive product ion spectra enabled isomers of unknown identity to be classified as 6-C or 8-C glycosides [25]. Combining retention time, CCS and accurate mass measurement (precursor and product ions), ‘known-unknowns’ enhanced specificity has been attained to facilitate authentication analysis and generate Passiflora species variant profiles, even though the precise nature of each component in molecular terms may not be known. Increased coverage and specificity have been achieved, where 255 isomer pairs were ion mobility separated and 86% of these components detected in only one of the Passiflora variants. The approach is beneficial where high purity reference standards are not available or are expensive. An example of species variant profiles separated by retention time and drift time are shown in Figure 8 for [M-H]- m/z 577.1563 isomers (C27H30O14). Identifying ‘known/unknowns’ and reassignment to ‘knowns’, can be achieved without reliance on analytical standards and has been described in detail elsewhere [19]. Flavonoid CCS libraries can be expanded where experimental CCS information has been compared with CCS prediction values and corresponding structural product ion information compared with historical profiling information. This strategy has been used to re-assign ‘known-unknown’ isomers to ‘knowns’ and expand the known Passiflora speciation fingerprint.
Conclusion
UPLC-IM-MS is a gateway to targeted specificity in non-targeted data independent screening assays. The evolution of data-independent analysis to profile medicinal plant species and complex analysis in itself, has been illustrated for the phytochemical profiling of four Passiflora species. Multifactor identification confidence is achieved using four identification points, comprised of precursor ion/product ion accurate mass, retention time and CCS measurements. As with accurate mass measurements, retention time, CCS measurements are seamlessly generated, to give rise to an additional specificity enhancing descriptor for all components in a sample.
It is advantageous to have the combined separation of ‘full scan’ UPLC-IM-MS. Data-independent HDMSE acquisition, facilitates generation of data akin to MS/MS, with production of cleaned up precursor/product ion spectra for all components in a sample. The necessity to set up targeted acquisitions and requirement to have specific prior knowledge of a sample’s components is removed.
Using four cumulative metrics precursor ion m/z, product ions m/z, tr, and CCS; a highly specific (non-targeted) ‘known–unknown’ species dependent fingerprint is created for phytochemical profiling. A vast amount of information is used that would conventionally remain redundant. The information obtained for the additional analyte ‘unknowns’, ‘unknown’ isomers, is used to populate ‘known–unknown’ databases. The strategy is used to extend and enhance speciation profiles, without the necessity to identify all species components. CCS prediction algorithms have evolved and been used to identify ‘known-unknowns’ transforming their assignment to ‘known’. In this research increased coverage and species specificity is achieved using UPLC-IM-MS, where 255 ion mobility separated isomer pairs of ‘known-unknowns’ were detected, of which 86% were specific to one Passiflora variant. Specificity in complex sample analysis is gained through ‘known’ and ‘known-unknown’ profiling.
Routine three-dimensional resolution using tr, m/z and IM drift time is used to separate, coeluting isobaric and isomeric components. IM as a matter of course provides separation (based on size, charge and shape) orthogonal to UPLC (hydrophobicity). Unachievable using accurate mass alone, chromatographically coeluting marker isomers have been differentiated using CCS and separation of distinctive positive/negative isomeric product ion ratios achieved.
UPLC afforded a time efficiency gain of 66% for analysis and greater peak capacity. High resolution mass spectrometry is routinely utilised at >20000-50000 FWHM. These resolution enhancements provide analytical flexibility through enhanced specificity of data independent acquisitions. Additionally, targeted data processing efficiency occurs as a consequence of informatics advances and incorporation of a CCS identification point [27].
1. Dawson JHJ and Guilhaus M. Orthogonal-acceleration time-of-flight mass spectrometer. Rapid Commun. Mass Spectrom. 1989; 3, 155.
2. Morris HR, Paxton T, Dell A, Langhorne J, Berg M, Bordoli RS, Hoyes J and Bateman RH. High Sensitivity Collisionally-activated Decomposition Tandem Mass Spectrometry on a Novel Quadrupole/Orthogonal-acceleration Time-of-flight Mass Spectrometer. Rapid Commun. Mass Spectrom. 1996; 10:889.
3. M Guilhaus, Mlynski V and Selby D. Perfect Timing: Time-of-flight Mass Spectrometry. Rapid Commun. Mass Spectrom. 1997; 11, 951–962.
4. Chernushevich IV, Loboda AV and Thomson BA. An introduction to quadrupole–time-of-flight mass spectrometry J. Mass Spectrom. 2001; 36, 849–865.
5. Swartz ME. UPLC™: An Introduction and Review, Journal of Liquid Chromatography & Related Technologies. 2005; 28:7-8, 1253-1263.
6. Harborne JB and Williams CA. Advances in flavonoid research since 1992. Phytochemistry. 2000; 55, 481–504.
7. Zeraik ML and Yariwake JH. Quantification of isoorientin and total flavonoids in Passiflora edulis fruit pulp by HPLC-UV/DAD. Microchemical Journal. 2010; 96, (1), 86-91.
8. Pereira CAM, Yariwake JH and McCullagh M. Distinction of the C-glycosylflavone isomer pairs orientin/isoorientin and vitexin/isovitexin using HPLC-MS exact mass measurement and in-source CID. Phytochem. Anal. 2005; 16, 295–301.
9. Giles K, Pringle SD, Worthington KR, Little D, Wildgoose J and Bateman RH. Applications of a travelling wave-based radio-frequency only stacked ring ion guide. Rapid Commun Mass Spectrom. 2004;18:2401-2414.
10. Pringle SD, Giles K and Wildgoose J. An investigation of the mobility separation of some peptide and protein ions using a new hybrid quadrupole/travelling wave IMS/oa-ToF instrument. Int J Mass Spectrom. 2007; 261(1):1-12.
11. D’Atrim V, Causon T, Hernandez-Alba O, Mutabazi A, Veuthey J, Cianferani S and Guillarme D. Adding a new separation dimension to MS and LC–MS: What is the utility of ion mobility spectrometry? J Sep Sci. 2017;1–48.
12. Eldrid C and Thalassinos K. Developments in tandem ion mobility mass spectrometry. Biochemical Society Transactions. 2020; 48(6):2457-2466.
13. Mason EA. and McDaniel EW. Transport Properties of Ions in Gases; 1988.
14. Dwivedi P, Puzon G, Tam M, Langlais D, Jackson S and Kaplan K. Metabolic Profiling of Escherichia coli by Ion Mobility-Mass Spectrometry with MALDI Ion Source. J. Mass Spectrom. 2010; 45 (12), 1383−1393.
15. Letertre M, Munjoma NC, Slade SE, Plumb RS, Swann J, Coen M, Nicholson JK and Wilson ID. Metabolic phenotyping using UPLC–MS and rapid microbore UPLC–IM–MS: Determination of the effect of different dietary regimes on the urinary metabolome of the Rat. Chromatographia. 2020; 83, 853–861.
16. McCullagh M, Giles K, Richardson K, Stead S and Palmer M. Investigations into the performance of travelling wave enabled conventional and cyclic ion mobility systems to characterise protomers of fluoroquinolone antibiotic residues. Rapid Commun Mass Spectrom. 2019;1–11.
17. Righetti L, Dreolin N, Celma A, McCullagh M, Barknowitz G, Sancho JV and Dall’Asta. C. Travelling Wave Ion Mobility-Derived Collision Cross Section for Mycotoxins: investigating interlaboratory and interplatform reproducibility. Agric. Food Chem. 2020; 68, 39, 10937–10943.
18. McCullagh M, Douce D, Van Hoeck E and Goscinny S. Exploring the Complexity of Steviol Glycosides Analysis Using Ion Mobility Mass Spectrometry. Anal. Chem. 2018; 90, 4585–4595.
19. McCullagh M, Goshawk J, Eatough D, Mortishire-Smith RJ, Pereira CAM, Yariwake JH and Vissers JPC. Profiling of the known-unknown Passiflora variant complement by liquid chromatography - Ion mobility - Mass spectrometry. 2021; Talanta 22, 121311.
20. Pereira CAM, Yariwake JH, Lanças FM, Wauters JN, Tits M and Angenot L. Comparison of HPTLC densitometric and HPLC-UV determination of flavonoids from Passiflora. Phytochem. Anal. 2004; 15, 241–248.
21. Qimin L, Heuvel VD, Delorenzo O, Corthout J, Pieters LC, Vlietinck AJ and Claeys M. Mass spectral characterization of C-glycosidic flavonoids isolated from a medicinal plant (Passiflora incarnata). J.Chromatogr. B. 1991; 562, 435–446.
22. Rehwald A, Meier B and Sticher O. Qualitative and quantitative reversed-phase high-performance liquid chromatography of flavonoids in Passiflora incarnata L. Pharm Helv. 1994; 69, 153–158.
23. Raffaelli A, Moneti G, Mercati V and Toja E. Mass spectrometric characterization of flavonoids in extracts from Passiflora incarnate. J.Chromatogr. A. 1997; 777, 223–231.
24. Farmacopéia Brasileira. 1977. (3rd edn). Atheneu: São Paulo.
25. McCullagh M, Pereira CAM and Yariwake JH. Use of ion mobility mass spectrometry to enhance cumulative analytical specificity and separation to profile 6-C/8-C-glycosylflavone critical isomer pairs and known–unknowns in medicinal plants. Phytochem. Anal. 2019; 30, 424–436.
26. McCullagh M, Douce D, Van Hoeck E and Goscinny S. Exploring the Complexity of Steviol Glycosides Analysis Using Ion Mobility Mass Spectrometry. Anal. Chem. 2018. 90, 4585−4595.
27. Goscinny S, McCullagh M, Far J, De Pauw E and Eppe G. Towards the use of ion mobility mass spectrometry-derived collision cross section as a screening approach for unambiguous identification of targeted pesticides in food. Rapid Commun Mass Spectrom. 2019;1–15.
28. Zhu MZ, Chen GL and Wu JL. Recent development in mass spectrometry and its hyphenated techniques for the analysis of medicinal plants. Phytochemical Analysis. 2018 29(4).
29. Cao JL, Wei JC, Chen MW, Su HX, Wan JB, Wang YT and Li P. Application of two-dimensional chromatography in the analysis of chinese herbal medicines. J Chromatogr A. 2014;1371:1-14.