HPLC, UHPLC
Published over 6 years ago. See the latest and most current information on HPLC, UHPLC.
Abstract: Chloramphenicol is one of the most frequently detected substances in the group of drugs banned from use in food producing animals. Therefore, chloramphenicol control is necessary in food of animal origin. We have developed a method that allows the determination of chloramphenicol in 22 different matrices using QuEChERS approaches with HPLC-MS/MS in negative electrospray mode. The method was successfully validated according European Union criteria.
Chloramphenicol (CAP) is a broad spectrum antibiotic that has been widely used since the 1950’s in livestock and has been successfully applied in all animal species. The recognised toxic effects of CAP for humans led to the restriction of its use in veterinary practice [1]. In 1994 the absence of safe residue levels of CAP, resulted in the European Union prohibiting its use for veterinary purposes in food producing animals, and no maximum residue limit (MRL) has been established for this antibiotic. Due to the ban on the use of this substance in the European Union, a limit of 0.3 μg kg−1 as a minimum required performance limit (MRPL) was set [2,3]. However, data from the Rapid Alert System for Food and Feed (RASFF) over the last 14 years has indicated that CAP contamination was present in various matrices with 488 notification events for CAP triggered, 442 for food and 46 for animal feed. The notifications listed concern a wide range of food products such as animal feed, water, milk, fish, honey, and meat. The origin of CAP in food of animal origin is a complex problem and depends on many factors. CAP is rapidly metabolised and is excreted in the urine as the main metabolite - glucuronide CAP (CAPG) [4]. Therefore, simple, sensitive and fast methods for determination of CAP and CAPG residues are needed. According to the literature, CAP residues may also be present in processed food, such as cooked products [5], therefore it can be assumed that preheated foods may also contain CAP residues. In addition, experience suggests that CAP can be transferred from milk to dairy products such as cottage cheese, cream, butter, and whey [6]. Also, according to the results of monitoring program for CAP, it can be found in products such as eggs, royal jelly, meat, milk, and dairy products [7]. This problem needed to be answered, and this article presents the method for the determination of CAP in twenty-two different matrices where it may be present and should be analysed.
The development of methods for the determination of residues of prohibited substances in many matrices (various types of biological material) is a multi-stage and complex process that must consider a wide variety of matrices such as fat, protein, water, sugars, and various food additives. All these different matrices can contain impurities which can lead to many overlapping peaks and a drifting background on mass spectrometry chromatograms [8]. In recent years, chromatographic methods for the determination of chloramphenicol have been developed, in various matrices based mainly on gas and liquid chromatography and using various forms of detection [4-6, 8-11]. Nevertheless, the development of a new atmospheric pressure ionisation technique in liquid chromatography coupled mass spectrometry detectors has contributed to the emergence of reliable techniques for the testing of chloramphenicol residues. These are now the most common methods of CAP determination [1,6,8,12-19], however there are only a few articles on the determination of CAP in a large number of matrices [1,6,8,12-17]. The purpose of this study was to create a simple, cheap and fast method with good recovery and good extract purification by employing the QuEChERS method for confirmation and quantification of CAP residues in 22 different matrices using the HPLC-MS/MS system, and validated in accordance with the criteria of Commission Decision 2002/657/EC [20].
2.1. Materials and Methods
Acetonitrile, isopropanol, and ethyl acetate were obtained from Merck (Darmstadt, Germany). Primary and secondary amine (PSA) was purchased from Supelco (Bellefonte, PA, USA). Sodium chloride (NaCl) and methanol were purchased from P.O.Ch. (Gliwice, Poland). CAP, CAP-D5 Anhydrous sodium sulphate (Na2SO4), anhydrous magnesium sulphate (MgSO4), and β-glucuronidase type HP-2 were procured from Sigma-Aldrich (St. Louis, MO, USA). Pre-heated magnesium sulphate (MgSO4) was prepared in our heating laboratory (400°C overnight). Octadecylsilane sorbent (C18), ammonium acetate, and acetic acid were obtained from J.T. Baker (Deventer, The Netherlands). All reagents were HPLC grade. Nanosep MF filter was supplied by Pall (Port Washington, NY, USA). Ultrapure water was filtered through a Millipore Milli-Q system (Burlington, MA, USA). Kinetex C8 column (75 mm x 2.1 mm x 2.6 μm) and C8 precolumn (4 mm × 2.1 mm × 4 μm) were obtained from Phenomenex (Torrance, CA, USA). Stock standard solution of CAP and CAP-D5 (1 mg mL−1) were prepared in methanol and stored in the dark in a 10 mL amber volumetric flask. This standard will remain stable for 12 months when stored at < −18°C. The working standard and internal standard solutions at the level of 0.01 μg mL−1 were prepared in methanol and stored in the dark in a 5 mL amber volumetric flask. This standard remains stable for 12 months when stored at <6°C. For the procedure optimisation and validation, milk, curd cheese, whey, butter, and sour cream with various protein and fat content, eggs, muscle (different species), liver, kidney, honey, sausage, ham, headcheese, intestines, aquaculture products (different species), royal jelly and mead were collected from supermarkets. The animal feed (for different species of livestock, such as pigs and poultry) were collected from a feed producer. Water, plasma, and urine (from different livestock species) were obtained. The samples have been checked for absence of CAP residues.
2.2. HPLC-MS/MS
The HPLC-MS/MS system was composed of an ABSciex ExionLC HPLC system (Concord, ON, Canada) connected to ABSciex API 5500 Qtrap mass spectrometer. The Analyst 1.6.3 software controlled the HPLC-MS/MS system and Multiquant 3.2 served to process the data. The MS system was operated in the electrospray negative ionisation mode with a capillary voltage of 4.5 kV. The multiplier was set at 1900 V. The desolvation temperature was set at 500°C, collision gas (N2)— 3.1 × 10e−5 torr; nebuliser gas (N2)—36 psi; gas 1 (air)—35 psi; gas 2 (air)—35 psi; curtain gas (N2)—36 psi. The chromatography was performed on a Kinetex C8 column, connected to a C8 precolumn. The mobile phase for LC analysis was composed of two solvents: A (0.5% isopropanol in 0.1% acetic acid in water) and B (methanol). Composition of mobile phase (A:B, v:v) was started at 15% of B to 2.5 min, then 45% at 3.0 min, and held for 3 min, then 15% and held for 3 min. The equilibration time was 3 min. The column was operated at 40°C at a flow rate of 0.4 mL min−1. The ions monitored by multiple reactions monitoring (MRM) were 321→194 and 321→152. The declustering potential (DP) was −105 eV. The optimised collision energy (CE) for CAP was −16 eV for the first product ion and −22 eV for the second one.
2.3. Sample Preparation
For urine, plasma, and homogenised liver, muscle and kidney, a 2 ± 0.05 g of the sample was mixed with 30 µL of the working solution internal standard (CAP-D5). Thereafter, 1.5 mL of 0.05 M acetate buffer, (pH 5.2) and 50 µl of β-glucuronidase were added. The sample was mixed for approx. 60 s and hydrolysed at a temperature of 50°C for 1 hr.
Intestine samples were first homogenised with liquid nitrogen and after that, 2 ± 0.05 g of the sample was mixed with 30 µL of the internal standard working solution (CAP-D5) and were mixed for 0.5 min on a vortex mixer at 349× rcf with 1.5 mL of water.
For other matrices a 2 ± 0.05 g homogenised sample with 30 µL of the internal standard working solution (CAP-D5) were mixed for 0.5 min on a vortex mixer at 349× rcf with 1.5 mL of water.
After this step all samples were mixed with 10 mL of acetonitrile (the optimum solvent as described in 3.2). Then, 0.5 g NaCl was added, and mixed for 1 min on a vortex mixer at 349× rcf and centrifuged at 2930× rcf for 10 min at about 6°C. Then, 7 mL of the top layer was transferred to a new centrifuged tube, and subsequently, 100 mg PSA, 200 mg C18 and 600 mg pre-heated anhydrous MgSO4 were added. This extract was then mixed for 2 min on a vortex at 349× rcf and centrifuged at 2930× rcf for 10 min at about 6°C. Then 5 mL of the top layer was transferred to a new centrifuge tube and evaporated under gentle nitrogen stream at about 45±5°C. The residue was re-dissolved in 0.3 mL of 0.5% isopropanol in 0.1% acetic acid, centrifuged with Nanosep MF filters (0.22 µm) at 9447× rcf at room temperature for 10 min and transferred to the autosampler vial for analysis.
2.4. Method validation
The method was validated according to the requirements of Commission Decision 2002/657/EC [3]. The validation parameters: selectivity, linearity, repeatability, within-laboratory reproducibility, recovery, uncertainty, decision limit (CCα), and detection capability (CCβ) were estimated [20-23]. In selectivity study, possible interferences encountered in the method have been checked by analysis of 20 blank samples, from different sources, for each matrix. The linearity was evaluated based on matrix-matched calibration curves, which were prepared by fortifying blank samples (for each matrix) at six con-centration levels corresponding to 0.1 (or 0.15 depending on the matrix); 0.3; 0.45; 0.6; 1.2; 2.4 μg kg−1 containing an internal standard (0.6 μg kg−1) [19]. The repeatability and reproducibility were determined at four concentration levels (six samples of each level) 0.1 (or 0.15 depending on the matrix); 0.3; 0.45; 0.6 μg kg−1. For matrices such as feed (pig, poultry), aquaculture (fish, shrimp) and muscle (chicken, pig), the samples in the validation were divided equally. For the purposes of repeatability, the samples were analysed by the same operators, on the same day with the same instrument and were calculated as the relative standard deviation (RSD, %). For within-laboratory reproducibility, two other sets of blank samples were fortified and analysed by different operators, on two different days with the same instrument, and were calculated as the relative standard deviation (RSD, %). The recovery was calculated by comparing the mean measured concentration with the fortified concentration of the samples [20-23].
3.1. HPLC-MS/MS Conditions
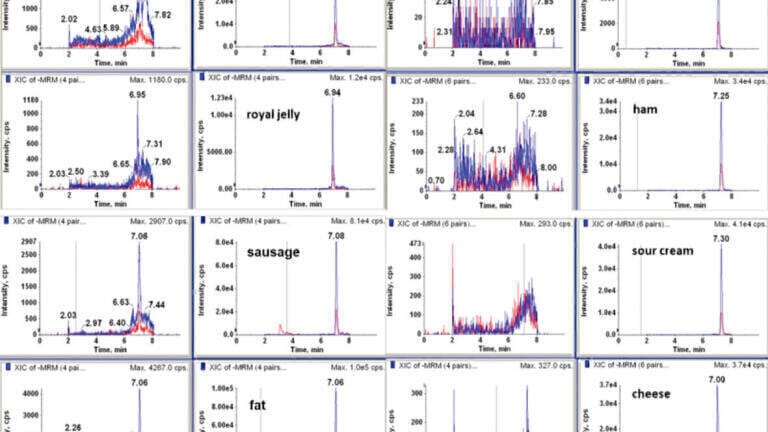
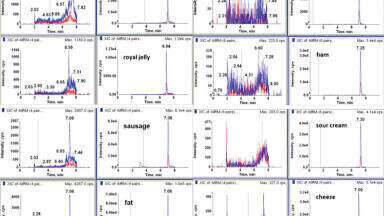
The best separation (symmetrical peak shape and minimal matrix effect) for CAP was achieved by using 0.5% isopropanol in 0.1% acetic acid in water: methanol. Examples of chromatograms of a blank and fortified samples of intestines, butter, curd cheese, plasma, honey, and urine samples at the level of 0.3 μg kg−1 are shown in Figure 1.
3.2. Optimisation of Sample Preparation
The determination of chloramphenicol may be divided into 1. the determination with a hydrolysis step (where CAP is mainly in a glucuronic form (liver, kidney, urine, plasma)) and 2. the direct determination of the parent drug (water, feed, milk and milk products, eggs, honey, royal jelly, muscle, and aquaculture products).
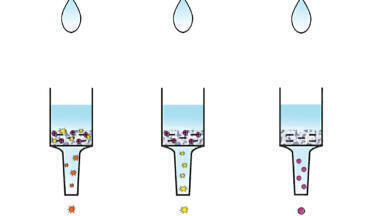
The most reproducible results for hydrolysis step, with the shortest time and at the lowest temperature, when no CAPG was confirmed, were achieved after 1 hour and at a temperature of at least 50°C. Many procedures describe the determination of CAP residues in biological matrices, such as honey, liver, muscle, kidney, milk and milk products, [1,6,13-17], but only a few are available for different kinds of food matrices of animal origin [1,6,9,13-19]. Based on our study we decided to use acetonitrile for CAP analysis, because it is a compromise between recovery (Figure 2) and purity of extracts (the purity of the extract was measured by the quantity of phospholipid interferences in the chromatogram after the extraction step), which is one of the most troublesome components causing difficulties in the analysis of biological samples for the determination of metabolites or xenobiotics by high-performance liquid chromatography especially when coupled with tandem mass spectrometry.
To compare the recovery, the samples were treated with acetonitrile, ethyl acetate, acetone, and chloroform:acetone (50:50), and then centrifuged. The best results (recovery) for all matrices were achieved for acetonitrile. Other mixtures such as ethyl acetate, acetone, and chloroform: acetone produced bad results for butter, cheese, feed and fat. The large amounts of co-extractive matrix compounds prevented evaporation to dryness. Poor results were also obtained for eggs with ethyl acetate (emulsion). Acetone: chloroform and acetone gave similarly poor recovery for other matrices (Figure 2).
For some matrices (muscle, aquaculture products, water, whey, liver, kidney, ham, headcheese, sausage, intestines, mead, royal jelly, honey, plasma, and urine) a better solution (recovery) could be ethyl acetate, but since the key objective was to simplify the procedure and to produce high purity extracts, it was decided to use acetonitrile for the extraction from all matrices. Other parameters for reagents (NaCl, C18 sorbent, PSA, anhydrous MgSO4) were selected based on our experience in selecting conditions for CAP determination in dairy products [6].
3.3. Method
Validation Results
The whole procedure was validated according to Decision 2002/657/EC on the quality standard [20]. The apparent recoveries were in the range of 93.1% to 108.0% with repeatability less than 9.2%, and within-laboratory reproducibility below 13.1%. The decision limit, and detection capability values were presented in Table 1.
The expanded uncertainty was calculated at the MRPL level [21] (Table 1). The calculation of ion suppression of the matrix effects for CAP for all matrices are below the suggested limit ±25%, which indicates that this is not a problem of this method [24] (Table 1). Values of correlation coefficients obtained from plotting the peak area corrected by internal standard in relation to the nominal concentration were higher than 0.98 at each matrix.
The validation study showed that, the presented method is a reliable confirmatory strategy for the determination of CAP and CAPG residue in all validated matrices. The detection limit and detection capability determined for CAP in twenty two matrices were below 0.3 μg kg−1 (MRPL). This simple method based on the QuEChERS and HPLC-MS/MS combination has illustrated that the total determination of CAP in various matrices based on the QuEChERS approach could be also successfully applied to many other matrices which were not verified during validation. This method is used in the National Monitoring Control Program in Poland.
1. M. Rejtharová, L. Rejthar, J. Chromatogr. A 1216, (2009), 8246–8253.
2. Commission Regulation (EC) No 1430/94 of 22 June 1994 amending Annexes I, II, III and IV of Council Regulation (EEC) No 2377/90 laying down a Community procedure for the establishment of maximum residue limits of veterinary medicinal products in foodstuffs of animal origin. 1994. Available online: https://ec.europa.eu/health/sites/health/files/files/mrl/regpdf/1994_06_22-1430_en.pdf (accessed on 30 November 2018).
3. Commission Decision (2003/181/EC) of 13 March 2003 amending Decision 2002/657/EC as regards the setting of minimum required performance limits (MRPLs) for certain residues in food of animal origin. Off. J. Eur. Union. L71, (2003), 17–18.
4. M. Gaugain, M. Chotard, D. Hurtaud-Pessel, E. Verdon, J. Chromatogr. B 1011, (2016), 145–150.
5. R.J. Shakila, S.A.P. Vyla, R. Saravana Kumar, G. Jeyasekaran, G.Indra Jasmine, Food Microbiol. 23, (2006), 47–51.
6. T. Sniegocki, M. Gbylik-Sikorska, A. Posyniak, Food Control. 57, (2015),411–418.
7. RASFF—the Rapid Alert System for Food and Feed. Available online: https://webgate.ec.europa.eu/rasff-window/portal/?event=search ResultList (accessed on 30 November 2018).
8. T. Sniegocki, B. Sell, M. Giergiel, A. Posyniak, Molecules. 24, (2019), 384.
9. P. J. Kijak, J AOAC Int 77, (1994), 34-40.
10. T. Nagata, H. Oka, J Agric Food Chem 44, (1996),1280-1284.
11. T. Sniegocki, A. Posyniak, J. Zmudzki, Bull Vet Inst Pulawy 50, (2006), 353–357.
12. T. Sniegocki, A. Posyniak, J. Zmudzki, Bull Vet Inst Pulawy 51, (2007), 59–64.
13. F. Barreto, C. Ribeiro, R. Barcellos, T. Dalla, J. Chromatogr. A 1449, (2016), 48–53.
14. L.R. Guidi, P.A.S. Tette, C. Fernandes, L.H.M. Silva, M.B.A. Gloria, Talanta 162, (2017), 324–338.
15. H.T. Rønning, K. Einarsen, T.N. Asp, J. Chromatogr. A. 1118, (2006), 226–233.
16. D. Rezende, N. Filho, G. Rocha, Food Addit. Contam. Part A. (2012), 37–41.
17. H. Tajik, H. Malekinejad, S.M. Razavi-Rouhani, M.R. Pajouhi, R. Mahmoudi, A. Haghnazari, Food Chem. Toxicol. 48, (2010), 2464–2468.
18. K. Kittler, W. Radeck, J. Polzer, Conf. Mater. EuroResidue VIII 23-26 May. (2016), 129–131.
19. Berendsen. B.J.; Zuidema, T.; de Jong, J.; Stolker, L.A.; Nielen, M.W. Anal Chim Acta 700, (2011), 78–85.
20. SANCO/2004/2726-rev 4 December 2008, Guidelines for the Implementaion of Decision 2002/657/EC. (2008). Available online: https://ec.europa.eu/food/sites/food/files/safety/docs/cs_vet-med-residues_cons_ 2004-2726rev4_en.pdf (accessed on 30 November 2018).
21. S.L.R. Ellison, A. Williams, Eurachem/CITAC guide: Quantifying Uncertainty in Analytical Measurement, Third edition, 2012 ISBN 978-0-948926-30-3. Available online: www.eurachem.org (accessed on 30 November 2018).
22. Capability of detection—Part 2: Methodology in the linear calibration case, This Br. Stand. Is UK Implement. ISO 11843-22000,\rincorporating Corrigendum Oct. 2007, Which Should Be Read Conjunction\rwith BS ISO 11843-12000. 2007 (2000) 1–2. Available online: https://www.iso.org/obp/ui/#iso:std:iso:11843:-2:ed-1:v1:en (accessed on 30 November 2018).
23. B.K. Matuszewski, M.L. Constanzer, Anal Chem. 1, (2003), 3019–3030.
24. M. Olejnik, P. Jedziniak, T. Szprengier-Juszkiewicz, J. Zmudzki, Rapid Commun. Mass Spectrom. 27, 2013, 437-442.





















-(1).jpg)