
Electrophoretic separations
Published over 13 years ago. See the latest and most current information on Electrophoretic separations.
The ability to retain and separate very polar, hydrophilic molecules in reversed-phase chromatography can be challenging and problematic. Retention may require the use of ion pair reagents, mobile phase pH modification, the use of concentrated buffers, or highly aqueous mobile phases. Such options have potential detrimental effects upon low-wavelength UV detection and reduced sensitivity in electrospray (ESI) mass spectrometry, and often still offer poor retention.
An example of such a challenging separation of very polar pharmaceutical analytes is shown in Figure 1. The methodology used to obtain this separation exhibited poor retention, with analytes eluting near the solvent front, and poor resolution of some components. Detection at very low wavelength was required, which limited the choice of the mobile phase and buffer. A range of columns, including polar end-capped phases, did not offer acceptable chromatography for the compounds of interest. A separation method was required for the analysis of this drug (analyte 4) in development at AstraZeneca, its degradation products, and the impurities arising from the synthetic process (analytes 1, 2, 3). Confidentiality prevents the disclosure of the name of the drug. Table 1 includes some of the physicochemical properties of these compounds. High pKa values show that the compounds are permanently charged in conventional HPLC conditions, and the negative log D values show that the compounds are extremely hydrophilic.
Retention mechanisms of polar compounds on porous graphitic carbon
The Hypercarb material has the ability to retain very polar compounds and has other unique properties as a stationary phase in HPLC [1-4]. Its chemical surface properties distinguish PGC from more conventional LC packings such as bonded-silica gels and polymers. PGC particles are spherical and fully porous with a porosity of approximately 75%. The surface of PGC is crystalline and highly reproducible with no micro pores. At the molecular level, PGC is made up of sheets of hexagonally arranged carbon atoms linked by the same conjugated 1.5-order bonds which are present in any large polynuclear aromatic hydrocarbon [3].
PGC behaves similarly to a strongly retentive alkyl-bonded silica gel for non-polar analytes; however its retention and selectivity behavior toward polar and structurally related compounds is very different. The retention of polar compounds can be explained by the polar retention effect (PREG – polar retention effect on graphite [3]) whereby solutes of increasing polarity showed a high affinity towards the graphite surface. With conventional alkyl-bonded silicas, the addition of a polar group to a molecule will normally reduce retention in the reversed-phase mode whereas with PGC retention is reduced to a much smaller extent or may even increase. Tanaka and co-workers [5] plotted log k for the various stationary phases against log P (where P is the octanol-water partition ratio). For C18 the correlation was very good, but this was not the case for PGC. The retention of polar compounds on PGC was much higher than expected, exhibiting k values 4 to 15 times higher than expected on the basis of their log P. This behaviour makes PGC well suited to the separation of very polar and ionised solutes such as carbohydrates and compounds with several hydroxyl, carboxyl, amino and other polar groups [6 -11].
The retention mechanism by graphite from aqueous / organic eluents is determined by the balance of three factors [3]:
a. Hydrophobicity effect, arising from resistance to the disruption of the structure of hydrogen-bonded solvents by non-hydrogen-bonding analytes. This is primarily a solution effect that tends to drive analytes out of solution.
b. Dispersive interactions of the London type between the graphite surface and the analytes. These are largely balanced by similar interactions between the graphite surface and the eluent (a) which is displaced by the analyte. Their net effect may either encourage or discourage retention, but they may have an important effect on selectivity.
c. Interaction of polarisable or polarised groups in the analyte with the polarisable surface of the graphite (Figure 2). These are additional to the normal dispersive interactions.
The overall effect of these competing interactions is that increasing the hydrophobicity of the analyte, for instance by adding alkyl groups into a molecule, always increases retention, as expected in a typical reversed-phase mode. However, increasing the polarity of the analyte by adding groups which can either donate or accept electrons or can polarise the graphite surface may also increase retention, particularly if these groups are constrained to be in close contact with the graphite surface. Therefore, the strength of interaction depends on both the molecular area of an analyte (and, therefore, shape of the analyte) in contact with the graphite surface and upon the nature and type of functional groups at the point of interaction with the flat graphite surface. The flatter the analyte, the closer its alignment to the graphite surface with a higher number of points of interaction possible, the greater the retention.
Goal
The purpose of the work described herein was to develop a methodology that can be effectively utilised for the retention of extremely polar compounds such as those described in Table 1. Porous graphitic carbon columns (Hypercarb) were found to provide suitable retention and separation of the analytes under analysis. The optimised method uses conventional LC mobile phase conditions that allow UV detection at a low wavelength of 195nm. Two versions of the method are described. The first is suitable for systems with low dwell volume (less than 100 µL), such as UHPLC equipment. This combination allows the use of narrow bore columns and fast gradients, resulting in 3× faster methods using approximately 11x less solvent. The second method was developed for systems with higher dwell volume (approximately 1100µL) and higher dead volume (such as those found in conventional HPLC equipment). This meant that a 4.6mm diameter column was essential to maintain an appropriate ratio between column volume and extra-column volume to minimise associated band-broadening. The second method also has two additional compounds (by-product 5 and component 6) added, which were incorporated as methodologies for different stages of manufacture were combined.
Experimental
Method development background
The application described herein entered development at AstraZeneca using a C18 column, whereby polar components were eluted isocratically under the starting conditions of 100mM potassium phosphate / acetonitrile (95:5, v/v) mobile phase [12]. A gradient section was present extending to 50% acetonitrile to ensure the elution of any late eluting impurities. Analysis time was 60 minutes per injection and exhibited poor retention of the newly identified polar components (analytes 2 and 3), as shown in Figure 1. Typically within AstraZeneca, new methods are developed through a process of screening a small range of orthogonal stationary phases, organic solvents and pH conditions. This identifies a suitable method in the majority of cases, often with little to no optimisation. Polar species are one area where this standardised approach to method development can fail to deliver a suitable method, as was the case in this application where no suitable methodology was identified under any of the conditions.
The separation in this application was hindered by the extent of the limitations presented by the analytes of interest. Poor UV chromophores necessitated detection at 195nm, reducing the number of available mobile phase options based on UV cut-off. Poor solution stability limited the range and concentration of pH modifiers that could be employed and poor solubility in non-aqueous solvents further limited the separation options.
Retention mechanisms involving HILIC, mixed-mode or normal phase approaches are typically used for the separation of polar species; these were all assessed but within the above restrictions could not be used to produce a suitably robust method.
The simplest next step approach was chosen: to assess the separation in 100% aqueous conditions. However, traditional C18 columns are not compatible with this mobile phase selection due to potential de-wetting/phase collapse [13]. Therefore, a polar embedded column that was compatible with 100% aqueous conditions was selected and assessed. A separation suitable to progress the projects development work was identified, (Figure 3), although upon extended use it became evident that the degree of resolution was not sufficient to produce a robust long term method and should impurities be present at elevated levels the separation obtained would no longer be baseline resolved which could lead to less accurate quantification.
At this stage a PGC column was investigated to assess whether this column could yield a suitably robust separation of the polar analytes under reverse phase conditions. The mobile phase composition was also investigated on the PGC column, and a higher buffer concentration was found to be required to ensure good peak shape and good resolution between peaks 3 and 4.
Method 1:
Test mix preparation
Approximately 3.00mg of compounds 1, 2, and 3 were dissolved in 100mL of sample diluents [water / acetonitrile (95:5, v/v)] to give a concentration of 0.03mg/mL impurity solution. Then 15.0mg of compound 4 was dissolved with 1mL impurity solution to give the final ratio of 0.2% impurity to parent compound (4).
Chromatographic method 1:
Instrument: Thermo Scientific™ Accela™ 600 and Open Autosampler
Column: Thermo Scientific Hypercarb™ 5µm, 100× 2.1mm
Mobile Phase:
A: 100 mM KH2P04 in LC-MS grade water
B: 100 mM KH2P04 : acetonitrile (LC-MS grade)( 50:50, v/v)
Gradient
conditions: T (min) % A %B
0.0 95 5
0.6 95 5
5.0 0 100
5.5 95 5
9.0 95 5
Flow rate: 400µL / min
UV: 195nm
Wash solvent: Water: Acetonitrile (95:5, v/v)
Injection volume: 1µL
Temperature: Ambient
Pressure (t=0): 252 bar
Method 2:
Test mix preparation:
Approximately 3.0mg of compounds 1, 2, 3, 5 and 6 were dissolved in 100mL of sample diluents diluents [water / acetonitrile (95:5 v/v)] to give a concentration of 0.03mg/mL impurity solution. Then, 1.5mg of compound 4 was dissolved in 1mL impurity solution to give the final ratio of 2.0% impurity to parent compound (4).
Chromatographic method 2:
Instrument: Conventional HPLC System
Column: Hypercarb™ 5µm, 100 × 4.6mm
Mobile Phase:
A: 100 mM KH2P04 in LC-MS grade Water
B: 100 mM KH2P04 : Acetonitrile (LC-MS grade) (50:50 v/v)
Gradient
conditions: T(min) % A %B
0.0 95 5
15.0 0 100
17.0 0 100
17.1 95 5
27.0 95 5
Flow rate: 1.50mL/min
UV: 195nm
Wash solvent: water: acetonitrile (95:5 v/v)
Injection volume: 5µL
Temperature: 60˚C
Pressure (t=0): 74 Bar
Results and discussion
Method 1
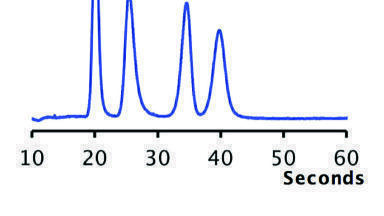
With the smaller ID column and fewer compounds, the analysis was carried out in less than 9 min (Figure 4). The peaks were eluted in less than 4 min and very high resolution could be achieved. To check the reproducibility of the method, over 40 runs were carried out. Some of the chromatograms are overlaid in Figure 5. The method is very robust and consistent. The RSD values on retention time from the 39 runs (excluding the first run) were calculated to be less than 0.2% for all four compounds. The results are recorded in Table 2.
Method 2
The need for a method suitable for use on a conventional HPLC system with high dwell volume and high dead volume required the use of a 4.6 mm ID column. Two additional compounds (analytes 5 and 6) were also added to the method requirements. The result of this was that the analysis had to be extended to 27 minutes. With this method, all of the peaks were separated with excellent resolution (Figure 6).
The method reproducibility was also established with 40 replicate injections and overlaid chromatograms of seven injections from through the run are shown in Figure 7. After the column equilibrated sufficiently, consistent results could be obtained.
Further method robustness and reproducibility was assessed by investigating column-to-column variability. Three columns with different batches of stationary phase were obtained and the application SST (containing analytes 1-6) was analysed on each. After each column had been suitably equilibrated each column produced equivalent chromatograms.
This method was subsequently validated and transferred to five other laboratories where equivalent chromatography was successfully demonstrated.
Conclusion
Porous graphitic carbon columns can be used successfully for the retention and separation of extremely polar compounds, which are problematic to retain in reversed-phase conditions. These columns offer a unique polar retention mechanism that often can be implemented simply, and can produce robust separations of extremely polar species under typical reversed phase conditions. In the example presented the methodology was successfully validated and transferred to other laboratories.
References
[1] H. Colin, G. Guiochon, J. Chromatogr., 158 (1978), 183
[2] J.H. Knox, K.K. Unger, H. Mueller, J. Liq. Chromatogr., 6 (1983), 1
[3] J.H. Knox, P. Ross, Adv. Chromatogr., 37 (1997), 73
[4] M. Melmer, T. Stangler, A. Premstaller, W. Lindner, J. Chrom. A, 1218 (2011) 118-123
[5] N. Tanaka, T. Tanigawa, K. Kimata, K. Hosoya, T. Araki, J. Chromatogr. A, 549 (1991) 29–41.
[6] C. West, C. Elfakir, M. Lafosse, J. Chrom. A, 1217 (2010) 3201-3216
[7] L. I. Monser, G. M. Greenway, Analytica Chimica Acta, 322 (1996) 63-68
[8] C. Antonio, T. Larson, A. Gilday, I. Graham, E. Bergström, J. Thomas-Oates, J. Chrom. A, 1172 (2007) 170-178
[9] K. Gaudin, T. Hanai, P. Chaminade, A. Baillet, J. Chrom. A, 1157 (2007) 56-64
[10] S. Bieri, E. Varesio, O. Muñoz, J-L. Veuthey, P. Christen, J Pharm. Biomed. Anal., 40 (2006) 545-551
[11] J. Vial, M-C. Hennion, A. Fernandez- Alba, A. Agüera, J. Chrom. A, Volume 937, 21-29, (2001)
[12] AstraZeneca Analytical Method Protocol
[13] Y.V. Kazakevich et al., J. Chromatogr. A, 913, 75–87 (2001).





















-(1).jpg)