
Bioanalytical
Published over 11 years ago. See the latest and most current information on Bioanalytical.
Abbreviations: PPGL – phaeochromocytoma and paraganglioma; PMets – plasma metadrenalines; UMets – urine metadrenalines; LC-MS/MS - liquid chromatography tandem mass spectrometry; NMA – normetadrenaline; MA – metadrenaline; 3-MT – 3-methoxytyramine; WCX – weak cation exchange; SCX – strong cation exchange; SPE – solid phase extraction; ACN – acetonitrile; MeOH – methanol; AmF – ammonium formate; AmAc – ammonium acetate; FA – formic acid; PFP – pentaflurophenyl; HILIC – hydrophobic interaction liquid chromatography; ECD – electrochemical detection
The biochemical investigation of phaeochromocytoma and paraganglioma (PPGL) has seen significant changes over the last fifty years. In large this has meant clinical laboratories have gone from using colorimetric assays for the measurement of adrenaline and noradrenaline, to the measurement of urinary catecholamines and metadrenalines and more latterly the measurement of plasma metadrenalines. Each approach has posed a number of analytical challenges to the clinical laboratorian. Herein is a critical review of the analytical methodologies available for measurement of urinary and plasma metadrenalines.
Introduction
PPGLs are a group of rare tumours that arise in large from chromaffin tissue from within the adrenal medulla (80-85%) and extra-adrenal sympathetic tissue in the head, neck and chest (10-20%) [1].
Paragangliomas can also be derived from the parasympathetic nervous system and as a consequence are biochemically silent (i.e. do not produce catecholamines).
PPGLs derived from the sympathetic nervous system typically produce the catecholamines adrenaline, noradrenaline and dopamine. It is the catecholamines that are responsible for the myriad of clinical features observed in patients with PPGL. Symptoms are often variable owing to the type, concentration and pattern of catecholamines produced by the PPGL.
Methods for the measurement of catecholamines date back to 1949 when the first colorimetric method for the measurement of adrenaline and noradrenaline was published [2]. Since then, there have been significant developments in the understanding of the pathophysiology of PPGL. This in combination with advances in analytical technologies available to measure catecholamines and their metabolites has meant that methodologies are now focused on the most clinically relevant analytes.
Several studies have shown [3-17] that the plasma and urinary methylated catecholamine metabolites (i.e. normetadrenaline (NMA), metadrenaline (MA)) have superior diagnostic performance characteristics (i.e. sensitivity and specificity) than plasma and urinary catecholamines for the diagnosis of PPGLs. Recent clinical practice guidelines [1] for the diagnosis of PPGLs state that total fractionated urine metadrenalines (UMets) or plasma free metadrenalines (PMets) should be used as an initial biochemical screen for PPGL. While there have been differences observed in the diagnostic sensitivity and specificity of urine and plasma metadrenalines using different analytical methodologies, to date there has not been a multi-centre head to head comparison study using liquid chromatography tandem mass spectrometry (LC-MS/MS) for analysis comparing the two. The latter is regarded as the gold standard method [1] for the measurement of metadrenalines.
This article will focus on analytical methods published for the measurement of urinary and plasma metadrenalines. For historical methods see the review published by Peaston et. al. [18]. It is no longer recommended [1] that plasma or urinary catecholamines are measured for the evaluation of a patient suspected to have a catecholamine secreting PPGL.
Urine Metadrenalines
Usually a 24h urine collection is required to assess MA and NMA output. MA and NMA to creatinine ratios have previously been reported [19], but are not widely used. Table 1 summarises methods published for the measurement of UMets [20-27]. Typically methods measure total (i.e. free and deconjugated) fractionated metadrenalines (i.e. NMA and MA separately). The majority of metadrenalines present in urine are conjugated with sulphate and glucuronide groups. The former resulting from a sulphotranseferase enzyme, SULT1A3 present in the gut. This means that the measurement of UMets requires cleavage of the sulphate and glucuronide groups from MA and NMA before analysis. This is usually achieved by acid hydrolysis and incubation at 100°C. It is also possible through incubation with a sulphatase and or glucuronidase, but is not done frequently. Few authors have published methods for the measurement of urinary free metadrenalines [22, 27] despite the theoretical improvement in diagnostic specificity they may provide.
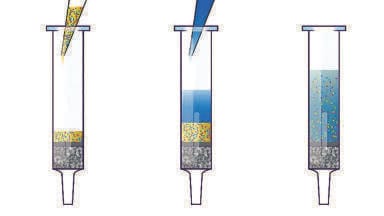
The majority of methods use off line [20-21, 23-27] solid phase extraction (SPE) [20-21, 23, 25-27] for the clean-up of metadrenalines. Several methods are based around cation exchange SPE [23, 25-27]. This approach is favoured as metadrenalines are weak bases (pKa ~9.5-10) and are thus highly suited to this approach. The mechanism underpinning this clean up exploits the interaction between MA and NMA when positively charged and the negatively charged sulphonic (strong cation exchange, SCX) or carboxcylic acid (weak cation exchange, WCX) functional residue on the cation exchange resin (Figure 1). Historical methods have combined ion exchange isolation with solvent extraction to improve specificity due to the complexity of chromatograms that early methods generated using only solid phase extraction. This is however time consuming and complex.
Recently Marrington et. al. [24] reported a dilution approach for sample preparation, which is simple and minimises sample preparation time. While this method did not appear to suffer from poor recovery or carry over, run times were increased. This approach can also reduce analytical column lifetime and increase the risk of instrument contamination.
Previous authors have used derivitisation for measurement of metadrenalines by gas chromatography mass spectrometry (GC-MS) [21] and fluorometric HPLC assays [28]. The former is analytically sound, but is not suited to a routine clinical laboratory as sample preparation can be complex and time consuming.
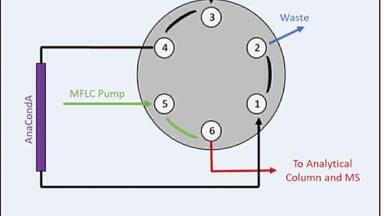
Some of the most convenient sample clean-up methods reported include the on-line Gilson ASTEDTM (automated sequential trace enrichment with dialysis) system coupled to high performance liquid chromatography electrochemical detection (HPLC-ECD). This system uses an in-line trace enrichment cartridge packed with sulphobutyl (HEMA-SB) packing material. In contrast the Gilson ASPEC (automated solid phase extraction for chromatography) utilises disposable pre-packed cartridges. Minimal sample preparation is required and acceptable analytical performance can be achieved using both approaches.
Davidson [22] reported the use of the former for the measurement of free urinary MA and NMA, but it has not gained widespread popularity, as limited data are available on the diagnostic performance characteristics of the free fraction of these analytes. This approach is particularly appealing as it provides a uniform approach for the measurement of urinary catecholamines and metadrenalines (Figure 2).
More recently Peitzsch et. al. [27] used off-line WCX SPE clean-up for the measurement of free metadrenalines by LC-MS/MS and reports that any diagnostic advantage of the free versus deconjugated metabolites (i.e. total) must be validated in a larger patient population, with additional estimates of diagnostic specificity in patients in whom tumours have been suspected and excluded.
A variety of HPLC columns have been used in published methods. In large the Atlantis T3 and HSS T3 columns have been used, owing in large, to their ability to chromatographically resolve the metadrenalines from the parent catecholamine compounds. This is in sharp contrast to PMets where hydrophilic interaction liquid chromatography (HILIC) columns have dominated, despite their reduced ability to resolve metadrenalines.
More recently, the pentaflurophenyl (PFP) column [24] demonstrated superior resolving power than HILIC and thus is highly suitable for the analysis of UMets.
As free and total metadrenalines are present at nanomolar to micromolar concentrations respectively, electrochemical detection (ECD) still remains the detection method of choice (UK NEQAS external quality assurance data distribution 178 (July 2014): 61/68 laboratories reported results using HPLC-ECD and 7/68 LC-MS/MS).
ECD is often the favoured detection method for UMets as it is more practical than gas chromatography [21] and fluorometric [28] procedures. ECD is highly suited to the measurement of UMets as they are easily oxidised and reduced, thus facilitating their quantification with a high degree of specificity in the presence of many other analytes in urine.
Despite this several studies have reported interferences in HPLC-ECD methods, these include paracetamol [29], labetalol, sulphasalazine, sotalol [30], buspirone [31], curry leaves [32] and amoxicillin [33]. However interferences can be minimised by optimising chromatographic and electrochemical conditions.
The increasing use of LC-MS/MS in the clinical laboratory has meant some laboratories are using mass spectrometry instead of ECD. This approach offers superior analytical specificity and reduced analytical run times, but is more expensive.
There have been reports of immunoassay being used for the measurement of UMets [14, 16], but are not discussed here as they are not commonly used in the clinical laboratory.
Plasma Metadrenalines
Although recent guidelines [1] recommend the measurement of PMets or UMets for the initial biochemical investigation for PPGL, the former are often preferred as they potentially offer greater diagnostic specificity. This is because they represent the increase in catecholamine metabolism by the tumour cells alone. As a consequence more recently method development in the field has focused on the measurement of PMets.
As the circulating concentrations of PMets are in the picomolar concentration range, sample clean-up is necessary due to the complexity of the plasma matrix. A variety of techniques have been investigated, with varying degrees of success. Off-line solid phase extraction, specifically WCX has been reported in the majority of studies [13, 35, 39, 40-41] (Table 2). The rationale for this approach is the same as that for UMets (Figure 1).
Marney et. al. [36] used a protein precipitation method in their investigation. This approach did not enrich or successfully eliminate major interfering matrix components from the sample as evidenced in poor analyte recoveries (35% and 66% NMA and MA, respectively) and a limit of quantification that was within the normal reference range.
More recently, significantly better recoveries of NMA and MA have been reported [13, 35, 39, 41] using WCX sample clean up. SCX SPE has also been investigated [37, 40], but did not show superior recoveries when compared to WCX.
As requests increase for the measurement of PMets in the clinical laboratory alternative approaches to sample clean up are being investigated. These include on-line sample clean-up. He et. al. [38] reported the use of a turboflow method, employing perfluoroheptanoic acid ion pairing agent in a water-acetonitrile mobile phase and a cyclone MCX-2 clean up column (strong cation exchange). This approach allows injection of plasma directly into the LC-MS/MS. This technology minimises method development time and analyst error, as well as providing excellent analytical sensitivity and specificity. This technology is however expensive.
Adaway et. al. [41] recently utilised the Waters online sample manager for the measurement of PMets, using the Oasis WCX cartridge. This method also benefits from reducing operator error, but offers no improvement in analytical specificity and sensitivity compared to off-line WCX SPE.
Most published methods for measurement of PMets use HILIC which, is highly suited to the measurement of PMets as they are very polar and thus enables one to exploit the properties offered by HILIC separation. HILIC is a variant of normal phase chromatography that provides a polar stationary phase coupled with an organic mobile phase. This results in chromatographic separation based on the differential distribution of the analyte between the organic mobile phase and the water layer that is adsorbed on to the HILIC solid phase [42]. The extent of retention on the analytical column is dependent on the polarity of the analyte. However, as discussed previously HILIC provides limited chromagraphic separation of PMets.
Twentyman et. al. [43] observed cross talk when using HILIC based separation. MA forms a product ion that has the same mass to charge ratio (m/z 151) as the precursor ion of 3-methoxytyramine (3-MT). This was reported to lead to falsely elevated 3-MT concentration, and as a consequence an erroneously high lower reference range.
Peitzsch et. al. [40] recently reported the use of an Acquity UPLC HSS T3 column which showed good retention of PMets, and optimal chromatographic separation compared to HILIC [13, 35, 39], thus minimising cross talk.
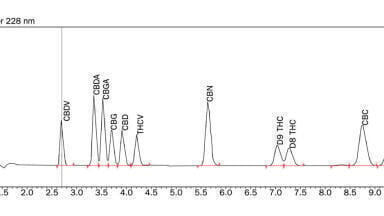
In-house investigations (unpublished) evaluated the use of a PFP column (Phenomenex, UK) for the separation of PMets (Figure 3). This provided optimal chromatographic separation, however it was observed that small changes in pH (~0.5 pH units) had a marked effect on retention time.
Additionally the authors of this article have looked at using a biphenyl column (Phenomenex, UK). The functional group on the biphenyl column appears to be less affected than the PFP column by small changes in pH and thus provides a more reproducible chromatography.
Early methods for the measurement of PMets used HPLC-ECD. However, the increased availability of LC-MS/MS in the clinical laboratory has led to more sensitive and specific methods being developed for PMets.
Peitzsch et. al. [40] recently reported that NMA and MA were measured on average up to 17% higher by LC-MS/MS than by HPLC-ECD. These findings are in agreement with previous reports [35] and are most likely explained by differences in internal standard correction in LC-MS/MS methods that utilise a stable deuterated internal standard.
Unlike the UMets, immunoassay has been more frequently utilised for the measurement of PMets in the clinical setting. A number of groups have investigated immunoassay as an alternative to HPLC-ECD and LC-MS/MS, as it is a low capital investment and is technically less demanding. However, this approach does not appear to offer the analytical sensitivity and specificity observed with LC-MS/MS [13-15]. Variable antibody cross-reactivity reduces the analytical specificity offered by immunoassay. Moreover immunoassays typically only measure the L-isomer of the NMA and MA. The latter may in part provides an explanation for the negative bias reported
by the RCPA external quality assurance scheme [44].
While LC-MS/MS provides a highly specific approach for the measurement of PMets, it is not without problems. Petteys et. al. [39] showed isoproterenol and MDMA interfered with the quantification of NMA, while isoetharine and MDA interfered with the quantification of MA using a LC-MS/MS method, however interferences can be minimised by the use of deuterated internal standards and optimisation of chromatographic conditions.
Conclusions
The measurement of UMets and PMets continues to be of interest to the clinical laboratory. Laboratories continue to investigate more sensitive and specific assays that are less labour intensive.
Manufacturers must work with laboratories to ensure that appropriate internal quality control material is made available to ensure the most accurate and reliable results are provided to clinicians investigating and managing patients with suspected PPGL.
References
1. Lenders JWM, Duh QY, Eisenhofer G, et. al.. J Clin Endocrinol Metab, 99 (2014) 1915–1942.
2. Von Euler US, Hamberg U. Science, 110 (1949) 561.
3. Lenders JW, Pacak K, Walther MM et. al.. JAMA, 287 (2002) 1427–1434.
4. Raber W, Raffesberg W, Bischof M, et. al.. Arch Intern Med, 160 (2000) 2957–2963.
5. Sawka AM, Jaeschke R, Singh RJ et. al.. J Clin Endocrinol Metab, 88 (2003) 553–558.
6. Unger N, Pitt C, Schmidt IL, et. al.. Eur J Endocrinol, 154 (2006) 409–417.
7. Giovanella L, Squin N, Ghelfo A, et. al.. QJ Nucl Med Mol Imaging, 50 (2006) 344–347.
8. Václavík J, Stejskal D, Lacnák B, et. al.. J Hypertens, 25 (2007) 1427–1431.
9. Gao YC, Lu HK, Luo QY, et. al.. Clin Exp Med, 8 (2008) 87–91.
10. Hickman PE, Leong M, Chang J, et. al.. Pathology, 41 (2009) 173–177.
11. Procopiou M, Finney H, Akker SA, et. al.. Eur J Endocrinol, 161 (2009) 131–140.
12. Grouzmann E, Drouard-Troalen L, Baudin E, et. al.. Eur J Endocrinol, 162 (2010) 951–960.
13. Peaston RT, Graham KS, Chambers E, et. al.. Clin Chim Acta, 411 (2010) 546–552.
14. Mullins F, O’Shea P, FitzGerald R, et. al.. Clin Chem Lab Med, 50 (2012)105–110.
15. Sarathi V, Pandit R, Jagtap V, et. al.. Endocr Pract, 17 (2011) 759–765.
16. Christensen TT, Frystyk J, Poulsen PL. Scand J Clin Lab Invest, 71 (2011) 695–700.
17. Unger N, Hinrichs J, Deutschbein T, et. al.. Exp Clin Endocrinol Diabetes, 120 (2012) 494–500.
18. Peaston RT, Weinkove C. Ann Clin Biochem, 41 (2004) 17–38.
19. Peaston RT, Lennard TW, Lai LC. J Clin Endocrinol Metab, 81 (1996) 1378-84.
20. Taylor RL, Singh RJ. Clin Chem, 48 (2002) 533–539.
21. Crockett DK, Frank EL, Roberts WL. Clin Chem, 48 (2002) 332-337.
22. Davidson DF. Ann Clin Biochem, 39 (2002) 557–566.
23. Whiting MJ. AnnClin Biochem, 46 (2009) 129–136.
24. Marrington R, Johnston J, Knowles S, et. al.. Ann Clin Biochem, 47 (2010) 467–475.
25. Clark ZD, Frank EL. Journal of Chrom. B Anal Tech Biomed Life Sci, 879 (2011) 3673-3680.
26. Gabler J, Miller A, Wang S. Clin Chem Lab Med, 49 (2011) 1213-1216.
27. Peitzsch M, Pelzel D, Glöckner S, et. al.. Clin Chim Acta, 418 (2013) 50–58.
28. Abeling NA, van Gennip AH, Overmars H, et. al.. Clin Chim Acta, 137 (1981) 211-226.
29. Davidson DF. Ann Clin Biochem, 41 (2004) 316-320.
30. Lenders JW, Eisenhofer G, Mannelli M, et. al.. Lancet, 366 (2005) 665-675.
31. Cook FJ, Chandler DW, Snyder DK. N Engl J Med (1995) 332:401.
32. Madahavaram H, Woolard GA. Ann Clin Biochem, 51 (2014) 400-405.
33. S. Barco, Alpigiani MG, Ghiggeri GM, et. al.. Clin Biochem, 47 (2014) 119–121.
34. Lagerstedt AS, O’Kane DJ, Singh RJ. Clin Chem, 50 (2004) 603–611.
35. De Jong WH, Graham KS, van der Molen JC, et. al.. Clin Chem, 53 (2007) 1684-1693.
36. Marney LC, Laha TJ, Baird GS, et. al.. Clin Chem, 54 (2008) 1729-1732.
37. Clarke MW, Cooke B, Hoad K, et. al.. Ann Clin Biochem, 48 (2011) 352-357.
38. He, X, Kozak, M. Anal Bioanal Chem, 402 (2012) 3003-3010.
39. Petteys BJ, Graham KS, Parnás ML et. al.. Clin Chim Acta, 413 (2012) 1459-1465.
40. Peitzsch M, Prejbisz A, Kroiß M, et. al.. Ann Clin Biochem, 50 (2013) 147-155.
41. Adaway JE, Peitzsch M, Keevil BG. Ann Clin Biochem, 2014 [e-pub]. DOI: 10.1177/0004563214546560.
42. Buszewski B, Noga S. Anal Bioanal Chem, 402 (2012) 231-247.
43. Twentyman JM, Cradic KW, Singh RJ et. al. Clin Chem, 5 (2012) 1156-1158.
44. Pillai D, Callen S. Ann Clin Biochem, 4(2010) 137-142.






















-(1).jpg)