This article discusses the development of chromatography modelling of the last 30 years from the first software package for calculating resolution and capacity factors to the visual modelling of chromatograms for testing peak movements with altering elution conditions.
Different approaches are discussed, such as retention modelling based on measurements, others based on molecular structure or on statistical considerations. The state-of-the-art will be demonstrated with a few applications of industrial importance.
Introduction
Computer supported chromatography method development [1,2,3] started around 1985 as IBM released the so called IBM PC, the first ’Personal Computer‘. The members of the project were Lloyd Snyder, John Dolan, Tom Jupille, founders of LCResources on the US West Coast and myself, who, after returning from Csaba Horváth’s Lab at Yale University founded the Institute for Applied Chromatography, located in Berlin-Kreuzberg on the 1st October 1981. The 4 of us decided to use the new technology of IBM-computers to write a program for HPLC method development. Jack Kirkland, a pioneer of HPLC at DuPont measured different properties on 1000 columns, and with Lloyd Snyder calculated the influence of the pore structure and ligand length in order to model band spreading. This was the beginning of ’DryLab‘, a name which Lloyd Snyder suggested for the software.
In 1988, the first iteration of the DryLab software was developed and allowed the modelling of band spreading, during optimisation of isocratic %B in DryLab I (I=isocratic) [4]. In 1989 modelling of gradient elution DryLab G (G=gradient) was developed [5]. First chromatograms for visualisation were plotted with *-characters. A few months later we were able to plot chromatograms for every change in experimental conditions. In the following years the software was further improved to isocratic multiparameter software, called ’DryLab Imp‘, where the user could model changes in pH, temperature, ionic strength, ternary eluent composition and ion-pair-chromatography. Gradient elution was more difficult to model with other factors, therefore the so called 2-dimensional modelling with gradient time (tG) and temperature (T) the (“tG-T-model”) was developed. At the same time a number of other factors, like column length, ID, particle size (dp), flow rate, dwell volume, gradient %Bstart and %Bend, and up to 10 gradient steps could be calculated. With these features DryLab was already in the 1990’s a multifactorial chromatography modelling software. The major feature of these models was their simplicity and visuality [6].
A very informative book on computer assisted method development was published in 1990 by Glajch and Snyder with 42 contributions to the theory and praxis of HPLC modelling [1] illustrating the work of leaders of the chromatographic scientific community working on separation predictions.
Some years later Sergej Galushko started his project, which he first named ’ChromDream’ which was later renamed ’ChromSword‘. The software allowed the prediction of retention time based on a compounds molecular structure which is important for those working in drug design [7,8]. To run the experiments he later introduced ’AutoChromSword‘ software which collected runs overnight in an automated fashion.
Other companies also introduced similar software packages. Agilent developed ICOS (intelligent computer optimization software) [9]. In France ’Osiris‘ was developed by the group of Heinisch, Rocca and Tschapla [10]. In Canada Mike McBrian introduced an optimisation software for chromatography with ACDLabs (Advanced Chromatography Development) [11]. During this time programs like ’Diamond’ and ’PESOS’ came and went. Around 2005 the company S-Matrix introduced “Fusion”, software which controlled Waters instruments to generate experiments and evaluate them according to statistical principles. This list is not complete and there were other software packages developed during this time, but these are beyond the scope of this article
Theory of RPC Modelling
Retention phenomena of reversed-phase chromatography (RPC) are described in many ways by different authors. The philosophy used in DryLab is described in the ’Solvophobic Theory‘ of Csaba Horváth, which was developed in the years 1975-1977 at Yale [12]. The fundamental concept of this theory is that retention in RPC is enforced by water, as the retarding component of the eluent. The uptake (dissolution) of nonpolar molecules in the water structure requires large amounts of energy. The retention factor k (also called the ’capacity factor‘) is proportional to the energy needed in this process. In the case of dibenzanthracene on a C8-phase, we find the following values for the capacity factor:
k in water kw ≈ 4000
k in acetonitrile kAN ≈ 1
Horváth and his team found that the only possible explanation for this extremely wide scale of retention times is the change in the surface tension of water altered by the addition of acetonitrile (AN) or methanol (MeOH). The strong lipophobicity of water can easily and continuously be reduced in this way, which is what occurs in gradient elution. Thus a typical approach to method development in RPC is to initially run a scouting gradient on a C18 column, which will typically resolves more than 95% of all compound peaks present in the sample.
Gradient elution typically starts with water or water-rich eluents. Upon injecting the sample into such a mobile phase (eluent), the water mixes with the hydrophobic sample components and forces them onto the surface of the C8 or C18 column packings. The capacity factors of organic molecules in water (Kw) are 103-106 times higher than in acetonitrile or methanol. By increasing the amount of the organic eluent, the retention force from water will become weaker, the surface tension of the eluent is reduced from 72 dyn/cm in water to approximately 22 dyn/cm at room temperature with a reduction in retention time occurring at the same time. This process has tremendous capabilities for separating complex mixtures in a highly reproducible manner for simultaneous qualitative and quantitative analysis.
In gradient elution, we can calculate the retention precisely for every component. Based on only two gradient runs, we can further calculate isocratic conditions and see how the k-values are reduced with increasing %B (percent organic) in the mobile phase.
The amazing ease of Reversed-Phase gradient elution is exhibited in the continuous reduction of the retention force of water by the increasing amount of the organic eluent (MeOH or AN). Fine differences in accessible solvophobic molecular surface areas, consisting of C-C, C-H and other nonpolar atomic bonds, combined with steps in the gradient, are sufficient to achieve reasonably good separations with almost any mixture in life science applications.
Modelling of Reversed-Phase separations is based on the measurement of both the retention time and the peak area [13,14]. The calculation of sample positions in the corresponding chromatograms in a Design of Experiments (DoE) enables the chromatographer to look at a small number of experiments in a virtual mode and generate a fast overview of separation choices. However by running a DoE, e.g. a tG-T model with 4 runs, we must realise that each chromatogram will look different. This however is the purpose of the exercise, as we want to learn how peaks move, so we can establish a model and can derive solutions for separation problems.
Experimental conditions
Column selection should be done carefully. We have a great number of RP-columns on the market. Snyder, Dolan, Carr, Engelhardt, Euerby, Tanaka and Petersson among others published excellent papers on column selectivity [15, 16] including more than 500 columns and demonstrated how to select the best columns for a separation. We used a YMC C18 120Å column, 150 x 4.6 mm, 5 µm (Waters, Milford, MA, USA) with a synthetic sample mixture developed for column testing at a flow rate of 2.0 mL/min. A Shimadzu Prominence (Shimadzu Europe, Duisburg, Germany) LC with dwell volume Vd: 0.4 mL and UV detection at 254 nm was used throughout the work. Modelling software was DryLab®4, v.4.0.10.15. (Molnar-Institute, Berlin, Germany). Eluent A was 0.025 M phosphate buffer at pH 2.8. Eluent B1 was acetonitrile (AN) Eluent B2 was methanol (MeOH) and a 50:50-mix of B1:B2. Gradient times were 20 and 60 min from 5 to 95% (B1+B2) at T1: 30 and T2: 60°C.
One and 2-dimensional models
If a chromatographer wants to understand peak movements caused by changes in experimental parameters, they must keep everything constant except one factor, like %B, tG or pH (or one factor at a time, OFAT). This helps to understand how a separation might change. Initially, this may appear to be an inefficient approach to spending time, however the opposite is true, since the chromatographer understands the separation better.
Changes of other parameters can also be modeled in DryLab by calculation: The influence of the flow rate, of the column length and ID, dwell volume, gradient start and end, steps, etc. So even an OFAT-model in DryLab allows the understanding of multifactorial changes. The most successful 2-dimensional model was and is still today the gradient time – temperature- or tG-T-model, especially when combined with a ternary gradient elution technique [17]. The tG-T-model which was used by Snyder in column characterisation [18], has lead to an extension into a 3D-resolution space, the Cube [14].
3-D-Models, the Cube
The first Cube model was demonstrated at the HPLC2009 conference in Dresden [14]. Soon afterwards, a number of papers appeared demonstrating the advantages of this new technology for industrial applications [19-23]. This new technology is especially well suited to improving the speed of older pharmacopoeial methods, as shown by Schmidt, where they reduced a method’s analysis times from 160 to 3 min, using DryLab and UHPLC [24].
The first step in this process is to plan a Design of Experiments (DoE) followed by the so called Peak Tracking process. The most efficient DoE is shown in Figure 1. (Figure 1).
Peak Tracking
PeakTracking is an important step in method development, as most chromatographers using a method are afraid of unexpected changes. Therefore small variations in working conditions should be carried out to test method robustness. The question is however, “How much should we vary a parameter?”.
If we change a parameter by very little, then we might not see hidden peaks. Therefore larger changes are required, e.g., two gradient times tG1 and tG2 with a factor 3 difference. In temperature optimization experiments we should have a difference 30-40°C and with pH, 0.6 pH units over 3 (or more) runs.
With these experiments we can create an experimental design with 4-12 runs, which is sufficient in most cases. We should learn as much as possible with the least possible number of runs.
It is widely accepted, that the so called tG-T-model is the best one to start with. It has only 4 runs and it allows simple peak tracking as shown in the following figures.
Initially the order of elution is established at the experimental points 2, 6 and 10 (see Figure 2).
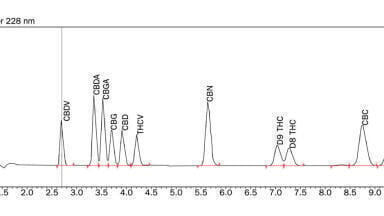
A peak tracking table of a tG-T-tC (tC = ternary eluent composition) model showing different elution profiles of the same mixture of 18 compounds in fewer than 12 different conditions [14]. The peak areas in those runs have a standard deviation of ca. 2% on average and can therefore efficiently be used to track moving peaks and establish robust conditions for routine applications.
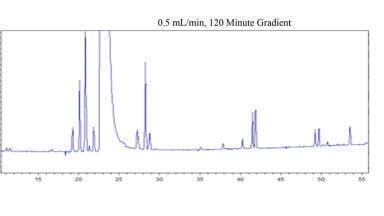
The next step is to align the 12 runs in the 3 tG-T-sheets. This is a process of looking at peak movements, peak overlaps and peak turnovers. Peak identification is based on peak areas, which represent the injected amount of the sample. Keeping this constant we get constant peak areas for a given compound in every run. Peak areas (concentration x volume = mass) are well suited to identify a peak. In peak overlaps the areas are additive. In Figure 3. we show the runs 1-2-3-4, where the organic eluent B1 is AN. Note the selectivity differences between the runs.
Then the peaks of the experiments 5-6-7-8 are aligned (Figure 4). Again there are different selectivities generated and several coeluting peak pairs observed.
At the end the last sheet of runs 9-10-11-12, which is the 100% methanol-sheet, all peaks are fully tracked (Figure 5.).
When peak tracking is complete, we then calculate between the 3 core sheets another 97 sheets, filling out the total space so we can simulate any chromatogram at any point in that whole space with more than 106 virtual chromatograms. The results are highly precise, up to 99.8% accuracy in retention times, which is comparable to the operational accuracies of most UHPLC instruments.
Method adjustments are much easier to implement when utilising resolution maps, as alterations of the “set point” or “working point” in the Design Space are not considered to be changes with post regulatory approval. This means, that changes in the Design Space (Figure 7) are possible without re-validation, allowing a much greater flexibility in the lab than in previous years.
From Figure 7 we can define several Design Spaces. The extension of the red areas (the possible Design Spaces) will give us a first idea about the robustness. We could also find suitable method parameter in methanol (front sheet of the cube in Figure 7 as well as in acetonitrile (back sheet in Figure 7).
From the design space as defined in Figure 7 we can get robustness information only for the measured parameters: Gradient time, pH and tC [%B2 in %B1] where B1 is acetonitrile and B2 is methanol. However, as DryLab®4 is able to calculate other changes which might occur at the same time, we can calculate the influence of additional parameters like flow rate or start- and end-%B of the gradient. No additional experiments are necessary for this kind of robustness calculation. The result is shown in Figure 8.
We can see in Figure 8. on the top of the graph the selected method parameter (tG = 46 min, T = 30°C and tC = 100 % MeOH as organic eluent) with estimated possible deviations from the nominal value. The temperature is assumed to deviate from the nominal value of 30°C by not more than +- 2°C, i.e. the true temperature is assumed to be in any experiment between 28 and 32°C). On the left graph the ’Frequency Distribution’ shows how often (N) a certain critical resolution (Rs,crit) occurs under any combination of possible, true parameter values. As can be seen from the graph, the failure rate, i.e., the number of experiments that could fall outside the required critical resolution Rs,crit =1.5, is = zero. That means that practically all experiments should fulfil the critical resolution requirement. The position of the “set point” or “working point” is of great importance, as many experiments cost enormous amount of resources. If the point is selected by trial and error, an analyst may have to change it and repeat a large number of experiments to find a new optimum. DryLab can calculate 6 experimental factors at 3 levels, i.e., 36 = 729 experiments in less than 1 minute!
The right graph in Figure 8 (‘Regression Coefficients’) describes the importance of each parameter, related to the selected deviation from the nominal value, for the critical resolution. As can be seen from the graph, temperature has the most important influence; a lower temperature gives a higher global resolution.
DryLab and the QbD movement
In 2002, the FDA instigated the development of the QbD concept which allowed more flexibility in industrial laboratories [14, 15]. DryLab demonstrated as early as 25 years ago, that systematic experimentation in HPLC is required.and has contributed to the development of Quality by Design in the analytical chemistry area. It was the first software demonstrating ’robust resolution maps‘, allowing the estimation of tolerance limits for every important parameter of a separation. DryLab is therefore an important tool to help meet QbD practices.
Method transfer
Method transfer is a problem in a global economy, where products travel over borders and are used in different location to generate the same analytical result. It is necessary to enhance this process using modelling software to ease the burden using virtual UHPLC models. This method transfer process is often instrument dependent and therefore it is important to understand how to utilise predictive software in method transfer. An example of successful method transfer using this approach is demonstrated in reference [23].
The so called Knowledge Management Protocol, which was discussed above in a short format, is a great help in dealing with regulatory authorities. In this way methods can be developed in an inspection-safe manner.
Economic considerations of modelling in reducing waste
In a steadily growing number of publications, the value and usability of retention modelling for fast and systematic method development has been demonstrated [17-22].
During the acetonitrile shortage, it was difficult to work in the HPLC lab as acetonitrile was not readily accessible. The development of the ternary Cube was important in this situation, and demonstrated that in most cases an alternative method utilising methanol could be implemented instead of acetonitrile [18]. We can contribute to a green chemistry by reducing waste through computer modelling and reduce our environmental impact by reducing the volume of mobile phase waste.
As we can see, there is not much difference between both methods, but the method in Figure 6 using MeOH as eluent B is more environmentally safe and is less expensive as the method in Figure 9 using AN as eluent B.
Conclusion
Methods with short analysis times can aid production of drugs faster and more economically than before, typically using UHPLC instrumentation. The use of modelling software allows the development of methods concordant with QbD criteria, increasing flexibility in routine operations. Retention and critical resolution problems can be more transparent than in the past. Method transfer is much easier using DryLab. Finally HPLC modelling is truly green as it saves time, energy and reduces waste.
References
1. J.L. Glajch and L.R. Snyder, eds, “Computer-Assisted Development for High Performance Liquid Chromatography”, Elsevier, Amsterdam, 1990 (J. Chromatogr., 485, 1989).
2. L.R. Snyder, J.J. Kirkland and J.L. Glajch, “Practical HPLC Method Development”, 1997, John Wiley & Sons, Inc
3. L.R.Snyder, J.W.Dolan, “High Performance Gradient Elution”, Wiley Interscience Hoboken, New Jersey, 2007
4. L.R.Snyder, J.W.Dolan, D.C.Lommen, J.Chromatogr., 485 (1989) 65-69.
5. J.W.Dolan, D.C.Lommen, L.R.Snyder, J.Chromatogr., 485 (1989) 91-112.
6. I. Molnár, K. Monks, From Csaba Horváth to Quality by Design: Visualizing Design Space in Selectivity Exploration of HPLC Separations, Chromatographia, 73 (2011) (Suppl.1) S5–S14.
7. S.V. Galushko, GIT Spezial Chromatographie, 2 (1996), 88–93.
8. S.V. Galushko, A.A. Kamenchuk and G.L. Pit, American Laboratory, 27 (1995) 421–432
9. A. Drouen et al., LC•GC, 9 (1991) 714.
10. S. Goga-Rémont, S. Heinisch, E. Lesellier, J. L. Rocca, A. Tschapla, Chromatographia, 51, (2000) 536
11. A. Bogomolov and M. McBrien , Mutual peak matching in a series of HPLC-DAD mixture analyses, Anal. Chim. Acta, 490 (2203)41–58
12. Cs. Horváth, W. Melander, I. Molnár, Solvophobic Interactions in Liquid Chromatography with Nonpolar Stationary Phases J. Chromatogr., 125 (1976) 129- 156.
13. I. Molnár, Computerized design of separation strategies by reversed-phase liquid chromatography: development of DryLab software, J. Chromatogr. A, 965 (2002) 175-194.
14. I. Molnár, H.-J. Rieger, K.E. Monks, Aspects of the „Design Space” in high pressure liquid chromatography method development, J. Chromatogr. A, 1217 (2010) 3193–3200.
15. L. R. Snyder, J. W. Dolan, P.W. Carr, The hydrophobic-subtraction model of reversed-phase column selectivity. J.Chromatogr. A, 1060 (2004) 77–116.
16. Melvin R. Euerby, Matthew James, Patrik Petersson, Practical implications of the “Tanaka” stationary phase characterization methodology using ultra high performance liquid chromatographic conditions, J.Chromatogr. A, 1228 (2012) 165– 174.
17. M. R. Euerby, F. Scannapieco, H.-J. Rieger, I. Molnár, Retention Modeling in Ternary Solvent Gradient Elution Reversed Phase Chromatography using 30 mm Columns, J.Chromatogr. A, 1121 (2006) 219-227.
18. J.W. Dolan, L.R. Snyder, T. Blanc, L.Van Heukelem, Selectivity Differences for C18 Reversed-Phase Columns as a Function of Temperature and Gradient Steepness. I. Optimizing Selectivity and Resolution, J.Chromatogr. A, 897 (2000) 37-50.
19. M. Euerby, G. Schad, H.-J. Rieger, I. Molnár, 3-Dimensional Retention Modelling of Gradient Time, Ternary Solvent-Strength and Temperature of the Reversed-phase Gradient Liquid Chromatography of a Complex Mixture of 22 Basic and Neutral Analytes using DryLab® 2010, Chromatography Today, Vol. 3, Dec. 2010, p.13.
20. Quality by Design: Multidimensional exploration of the design space in high performance liquid chromatography method development for better robustness before validation, K.Monks, I.Molnár, H.J.Rieger, B.Bogáti, E.Szabó, J.Chromatogr. A, 1232 (2012) 218-230.
21. A stepwise strategy employing automated screening and DryLab modeling for the development of robust methods for challenging high performance liquid chromatography separations: a case study, K. Jayaraman, A.J. Alexander, Y. Hu, F.P. Tomasella, Anal. Chim. Acta, 696 [1-2] (2011) 116-124.
22. Rapid high performance liquid chromatography method development with high prediction accuracy, using 5 cm long narrow bore columns packed with sub-2_m particles and Design Space computer modeling Sz. Fekete, J. Fekete, I. Molnár, K. Ganzler, J. Chromatogr. A, 1216 7816-7823 (2009).
23. I. Molnár, K.E. Monks, H.-J. Rieger, B.-T. Erxleben, LCGC-Magazine, Experimental Combination of Method Development Strategies in a Working Environment of Different Instrumental Set-ups, 7, 5 (2011), 2–8.
24. A. Schmidt, I. Molnár, Using an innovative Quality-by-Design approach for development of a stability indicating UPLC method for ebastine in the API and pharmaceutical formulations,
J. Pharm.Biomed.Anal., accepted for publication.






















-(1).jpg)