Bioanalytical
Published over 5 years ago. See the latest and most current information on Bioanalytical.
In the separation of species which are structurally similar, such as the impurities of a drug compound, the use of shallow gradients is often beneficial. Shallow gradients can be obtained either by increasing the gradient time or the flow rate. The former is generally used as increasing the flow rate can result in excessive pressures and peak broadening. However, under the right circumstances, the alternative of using high flow rates becomes feasible, and this is quite beneficial as it allows the shallow gradient to be obtained with a shorter run time. In this work, a column packed with fused core particles of 2.7 µm diameter, run at elevated temperature, was used to generate a high-flow-rate-induced shallow gradient. The use of elevated temperatures typically results in a significant reduction of any shape selectivity that may be offered by the column. This is unfortunate as shape selectivity can also be quite valuable in the separation of related substances. In this work, we show that by use of columns containing a biphenyl stationary phase it was possible to maintain a reasonable degree of shape selectivity at elevated temperatures. Therefore, it was possible to improve the separation by reducing the steepness of the gradient without increasing the run time; and simultaneously, obtain a reasonable degree of shape selectivity. We argue that this would be less feasible with sub-2-um columns and hence the use of ultra-high pressure liquid chromatography may be overvalued in cases where shallow gradients are desired.
In our laboratory we frequently develop methods for the quantitation of degradation products or process impurities of an active pharmaceutical ingredient. Such methods must be capable of separating compounds which are very similar in size, structure, and polarity. It has been our experience that methods which utilise very shallow gradients and which offer a degree of shape selectivity can be particularly beneficial in this regard. The shallower gradient can be accomplished by increasing either the gradient time or the flow rate. The latter is preferable if a shorter run time is desired. In this work, we evaluated columns packed with 2.7 µm fused-core particles, run at elevated temperatures, to allow shallow gradients to be obtained in less time. Furthermore, the use of a biphenyl stationary phase made it possible to, simultaneously, obtain a degree of shape selectivity in the separation, despite the elevated temperature. We tested this approach using one of the more challenging samples we have recently encountered in our laboratory.
The significance of gradient steepness has been addressed in the literature [1-6] including at elevated temperatures [7]. From the standpoint of improving resolution, a specific definition of gradient steepness is relevant, which can be defined as the gradient rate (Δɸ/tG) divided by the linear velocity of the mobile phase. Mathematically speaking, we could express this as per relationship (1), where Δɸ is the fractional change in mobile phase strength during the gradient, tG is the time over which the gradient is executed, F is the mobile phase flow rate, and dc is the inner diameter of the chromatographic column.
It may be noted that this is very similar to gradient steepness as defined by linear solvent strength theory [6], presented as relationship (2); where L is the length of the column, S is a constant which increases with the molecular weight of the analyte, ε is the interparticle porosity, and other terms are as defined above.
The difference is that in relationship (1) column length has been removed and the constant terms have been dropped so as to express a simple proportionality. The column length term was removed as a reduction in column length would not be beneficial for the present purpose, and could potentially compromise the separation [8-10].
From equation (1), it may be thought that reduction of Δɸ could be another approach to reducing the steepness of a gradient. However, this approach is generally not ideal as reductions in Δɸ may also have a negative effect on the separation. In this work, modifications to Δɸ will be made for a different purpose, which is to maintain an equivalent mobile phase elution strength when the column temperature is changed [11-12].
Considering the options of increasing the gradient time (tG) or the flow rate (F), the latter approach is preferable as it would accomplish the shallower gradient without increasing the run time. Surprisingly, to our knowledge, there are very few examples in the literature of the deliberate use of this approach. What is required is the ability to increase the flow rate significantly without going beyond the usable pressure range and/or creating detrimental viscous heating issues [13-17] or band broadening due to kinetic effects [18-20]. With conventional HPLC the ability to do this is limited as we will quickly encounter any or all of these difficulties. Mutton utilised high flow rates to preserve gradient shallowness with conventional HPLC (particles sizes of 3 µm and 5 µm); however, the column length was reduced by a factor of 3 in this work and the result was a reduction of run time but with a loss of resolution [21].
It may seem that UHPLC would be a good option for this approach, as the use of smaller particles leads to lower plate heights even when working at linear velocities above the optimal [22-29] and because UHPLC systems are designed to work at higher pressures. However, in order to accomplish a significant reduction of the gradient steepness, a fairly substantial increase in flow rate would be necessary. And this, in combination with sub 2 µm particles, would generally put the pressure out of range of most UHPLC systems, unless using significantly elevated temperatures. For example, it was calculated that if working at the commonly used temperature of 30°C, with the column used in this study but with the particle size reduced to 1.7 µm, and with the flow rate increased by a factor of
three, the resulting pressure would be 1560 bar, which is beyond on the range of today’s commercial UHPLC systems. Thus, we would have difficulties working at this pressure and would likely observe viscous heating issues as well. Shorter columns would offset this to some extent; however, this could be detrimental to the separation, as mentioned above.
There are three modes of liquid chromatography that generally allow for higher optimal linear velocities and flatter van Deemter curves, without generating excessive pressures: High temperature HPLC [12,30-37], the use of columns packed with fused core particles [38-44], and monolithic columns [45-49]. When these techniques are used in combination, even flatter van Deemter curves would be expected. The use of high temperatures and flow rates have previously been evaluated for both fused-core [50-54] and monolithic columns [55-58]; and both column formats have previously been used for separation of related substances [49,59]. In this work, we evaluated a 2.7 µm fused core column at an elevated temperature of 80°C, with regards to the ability to obtain shallower gradients by increasing the flow rate, and therefore, without increasing the run time.
As mentioned above, the use of a method which offers some shape selectivity can also be quite beneficial in the separation of related substances, and this is particularly so when a portion of the molecular structure is planar (which is quite common for pharmaceuticals). It has been demonstrated in the literature that the degree of shape selectivity observed typically decreases as temperature increases [60-64]. A biphenyl phase was used in this work as it is known to offer shape selectivity [65-71], and previous experience in our laboratory (unpublished) has led us to believe that the shape selectivity offered by this phase would be less effected by temperature. Another unique feature of biphenyl columns is that they contribute both π-π and hydrogen bonding interactions which can often be helpful in the separation of molecules with aromatic rings, as in the present work.
All chemicals used for the study were reagent grade or better. Mobile phase A consisted of 99.0% water, 0.6% acetonitrile, and 0.4% tetrahydrofuran. Mobile phase B consisted of 67% acetonitrile and 33% tetrahydrofuran. Approximately 0.1% formic acid was added to both mobile phases. The sample used for all experiments consisted of a pharmaceutical formulation that had been aged for 6 months at 40°C. The sample was prepared by weighing 2.5 g of drug product into a 5 mL flask and bringing to final volume with methanol containing 0.1% formic acid, resulting in a concentration of 25 mg/mL for the active ingredient. The structure of the active ingredient is proprietary; however its empirical formula is C16H13F2N3O3, it has a molecular weight of 333 g/mole, contains several rings within its structure, and a portion of the molecule is planar. An aged sample was used deliberately so as to ensure the presence of related substances. The column used in this study was a Raptor biphenyl purchased from Restek, 3x100 mm, 2.7 µm.
An Agilent 1200 HPLC system was used, with a G1312A Binary Pump, a G1315B Diode Array Detector, and a G1329A Automatic Sampler (Agilent Technologies, Wilmington, DE, USA). All runs were made with a 10 µL injection and data were collected at a wavelength of 301 nm.
Several minor modifications were made, to the HPLC system, to ensure optimal results when working at elevated temperatures. First, the system was plumbed such that the mobile phase went through both sides of the heating block (whereas typically only one side is used) and an extra length of tubing, of dimensions 500 mm x 0.17 mm ID, was placed between the heating block and the column in order to ensure that the mobile phase had sufficient time to pre-heat, particularly given the fairly high flow rates that were used in these experiments. Additionally, an extra length of tubing, of dimensions 600 mm x 0.17 mm ID, was placed between the column and the detector, with approximately 100 mm of this tubing placed into a 400 mL beaker filled with water, in order to cool the mobile phase prior to reaching the detector. Lastly, a 100 psi back pressure regulator obtained from Upchurch Scientific® was placed downstream of the detector to prevent any boiling of the mobile phase.
The quality of the separations conducted in this study were evaluated with respect to run time and with the metric of peak capacity, values of which were calculated as per equation (3); where tr,last and tr,first are the retention times of the last eluting and first eluting peak of interest, respectively, F is the mobile phase flow rate, and Waverage is the average peak width of the related substance peaks being separated. This relationship has previously been used in the literature [1,3,5,7 72-74] and provides an effective way of expressing the separation power of a gradient method.
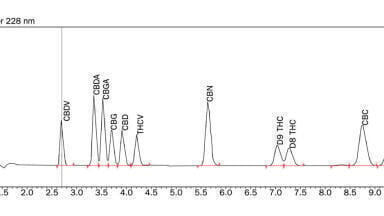
For the purpose of these calculations, the run time was taken as the retention time of the last peak of interest (the peak eluting just after 17 minutes in Figure 1a). The parameters used for these experiments, as well as the steepness of each gradient, are summarised in Table 1. The peak capacity and the run times are summarised in Table 2.
The starting point, for this study, is the chromatogram obtained with the column at 80°C, with a flow rate of 0.5 mL/min, and a gradient time of 26.7 minutes. This is presented in Figure 1a (note that the time axis is expanded in the figures so as to make the peaks of interest easy to visualise). It can be seen from the chromatogram that this separation was less than optimal as many of the peaks are not well resolved. The peak capacity was calculated to be 51. Figure 1b shows the result obtained under the same conditions, except that the gradient time was increased by a factor of four, thereby reducing the gradient steepness by the same factor. The resolution obtained is now visibly improved, particularly for the peaks which elute subsequent to the active ingredient (the large peak), such that the peaks are generally baseline resolved. The peak capacity, under these conditions, increased to 89. However, the run time was now longer by a factor of 2.6. The changes observed in the peaks eluting before the active ingredient are believed to be due to changes in selectivity which can occur, in gradient methods, when either the gradient time or flow rate are changed [6].
Figure 1c shows the result obtained when the flow rate was increased by a factor of four instead of the gradient time, resulting in a gradient steepness which is equivalent to what was used for the chromatogram of Figure 1b. The selectivity (peak spacing) of the chromatography is very similar to what was observed in Figure 1b; thus, demonstrating that the same gradient steepness has been obtained. The run time in Figure 1c was four times faster than the run of Figure 1b; and, in fact, was actually 6 minutes shorter than the original run (Figure 1a). However, some band broadening has clearly developed in this run which suggests that, despite the use of a fused-core particle at elevated temperature, the flow rate of 2.0 mL/min was too high and has resulted in a loss of efficiency. The peak capacity was determined to be 54. Hence, this run was very similar to Figure 1a, both in terms of separation power and speed. It seems that the benefit of the improved peak separation due to the shallower gradient was almost exactly cancelled by the loss of efficiency due to the higher flow rate.
Given that the somewhat disappointing separation obtained at 2 mL/min was believed to be due to setting the flow rate too high, an additional experiment was run where the flow was reduced to 1.5 mL/min and the gradient time was increased to 35.6 minutes. In this way, the same gradient steepness was obtained, but the shallower gradient was achieved by a combination of increasing the flow rate by a factor of three and increasing the gradient time by a factor of 1.33. The result, shown in Figure 1d, was that the same selectivity was again seen but now with better efficiency. In comparison to the initial steeper-gradient run (Figure 1a) a better separation has been obtained, as the peak capacity increased from 51 to 70 (a 37% improvement), but with essentially no change in run time (in fact the run time was two minutes shorter). Therefore, the goal of improving the separation power without increasing the run time has been achieved.
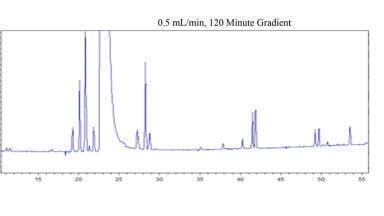
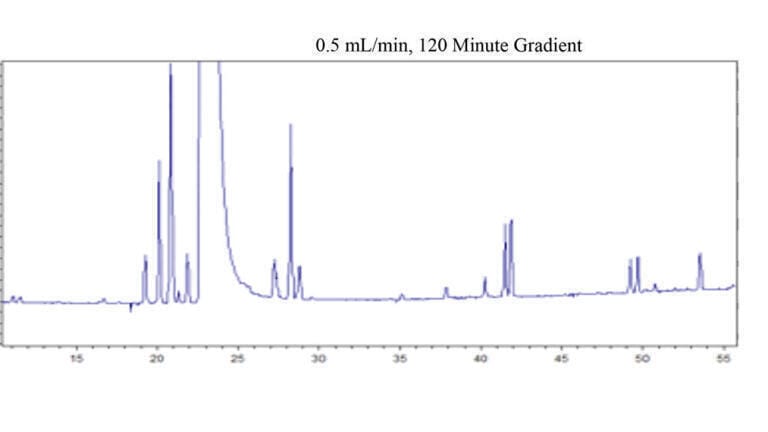
In order to evaluate the effect of temperature, the chromatogram depicted in Figure 2 was run with conditions similar to Figure 1b, but with the temperature set to 30°C. When using acetonitrile based mobile phases, it has been suggested that the acetonitrile content should be increased by 1% for every 5°C reduction in temperature to maintain equivalent elution strength [11]. Therefore, the high end of the gradient was set to 95% mobile phase B instead of 85%. The gradient time was then adjusted to 120 minutes so that the steepness of the gradient would be equivalent to that of experiment 1b. In this way, an effort was made such that temperature would be the only variable changing between the runs shown in Figure 1b and Figure 2.
While not identical, the chromatograms of Figure 1b and Figure 2 are very similar for the peaks which elute subsequent to the active ingredient. The differences seen in the earlier part of the chromatogram are believed to be due to changes in selectivity and elution order, as a function of the different temperature. The peak capacity was 103, which is only moderately better than what was observed for Figure 1b. The similar quality of the separation at 30°C, compared to 80°C (other than the noted exception), provides support for the contention that the shape selectivity of biphenyl phases are less temperature dependent than what has generally been reported for most columns. It is most likely the ‘slotted’ nature of these stationary phases (i.e. consisting of ligands that are fairly rigid, with room in between the ligands where the analytes can interact) that is responsible for the lesser dependence of shape selectivity on temperature. However, this is speculative and further studies are warranted to better understand the shape selectivity of these phases and the effect of temperature.
Experiments were conducted to evaluate the expectation, from theory, that reducing the steepness of a chromatographic gradient can be accomplished either by increasing the gradient time or by increasing the flow rate, and that the effect on selectivity (or peak spacing) should be essentially equivalent. It was expected that fused core columns, run at elevated temperatures, would be ideal for this purpose as they allow increased flow rates to be used without generating excessive pressures or peak broadening.
Our data generally support the validity of this approach, as similar selectivity was obtained by virtue of elevating the gradient time or the flow rate, but with the run time being faster when using the high-flow-rate alternative. It was found that increasing the flow rate to 2.0 mL/min was too high resulting in a loss of efficiency, which largely cancelled the benefits of the shallower gradient. When the experiment was run with the flow rate reduced to 1.5 mL/min, and the gradient time increased, so as to maintain the same gradient steepness, the peak capacity was 37% larger than that of the original run and with the analysis time actually reduced by two minutes. Thus, the goal of improving the separation by use of a shallower gradient, without increasing the run time was achieved.
Our data suggest that, in addition to the use of a somewhat more moderate flow rate, moderately smaller particle sizes, perhaps in the range of 2.0 to 2.4 µm, would be more optimal. However, particle sizes much smaller than this (i.e. UHPLC columns) are not believed to be ideal as the ability to increase the flow rate significantly becomes limited, unless using very high temperatures. It may also be noted that techniques such as supercritical fluid chromatography, HILIC, and classical normal phase chromatography would lend themselves particularly well to this approach due to the inherently lower pressures that are typically observed; and, in these cases, sub-2-µm particles would be feasible.
Our data also provide support for the hypothesis that biphenyl stationary phases offer a shape selectivity which is less effected by temperature than what has been reported, for other columns, in the literature. Further studies should be run to confirm this and to better understand the nature of the shape selectivity offered by these phases.
Lastly, we note that recent approaches which are used for comparison of chromatographic methods typically evaluate the combination of kinetic factors effecting band broadening, linear velocity, and the maximum column length that can be used given the pressure tolerance of the method. The additional variable of the steepness of the gradient is generally not considered but perhaps should be, given its importance.
1. Neue, U.D.; Carmody, J.L.; Cheng, Y.F.; Lu, Z.; Phoebe, C.H.; Wheat, T.E. and Editors, Chapter 3, Design of Rapid Gradient Method for the Analysis of Combinatorial Chemistry Libraries and the Preparation of Pure Compounds, in Advances in Chromatography, Volume 41, CRC Press Taylor and Francis Group, 2001.
2. Neue, U.D.; Alden, B.A.; Iraneta, P.C.; Mendez, A.; Grumbach, E.S.; Tran, K.; Diehl, D.M. and Editors, Chapter 4, HPLC Columns for Pharmaceutical Analysis, in Handbook of Pharmaceutical Analysis by HPLC, Volume 6, Elsevier Academic Press, 2005.
3. Neue, U.D.; Cheng, Y.F.; Lu, Z. and Editors, Chapter 1.2, Fast Gradient Separations, in HPLC Made to Measure: A Practical Handbook for Optimization, Wiley-VCH, 2006.
4. Cheng, Y.F.; Lu, Z.; Neue, U.D. Ultrafast liquid chromatography/ultraviolet and liquid chromatography/tandem mass spectrometric analysis, Rapid Commun. Mass Spectrom., 2001, 15, 141-151.
5. Neue, U.D. Theory of peak capacity in gradient elution, J. Chromatogr. A, 2005, 1079, 153-161.
6. Snyder, L.R.; Dolan, J.W. High Performance Gradient Elution: The Practical Application of the Linear-Solvent-Strength Model, Wiley, 2007.
7. Neue, U.D.; Mazzeo, J.R. A theoretical study of the optimization of gradients at elevated temperatures, J. Sep. Sci., 2001, 24, 921-929.
8. Engelhardt, H.; Elgass, H. Optimization of gradient elution, separation of fatty acid phenacyl esters, J. Chromatogr. A, 1978, 158, 249-259.
9. Jandera, P.; Churacek, J. Gradient Elution in Column Liquid Chromatography, Elsevier, 1985.
10. Dolan, J.W.; Snyder, L.R. Maintaining fixed band spacing when changing column dimensions in gradient elution, J. Chromatogr. A, 1998, 799, 21-34.
11. Chen, M.H.; Horvath, C. Temperature programming and gradient elution in reversed-phase chromatography with packed capillary columns, J. Chromatogr. A, 1997, 788, 51-61.
12. Teutenberg, T. High-Temperature Liquid Chromatography: A User’s Guide for Method Development, RSC Publishing, 2010.
13. Poppe, H.; Kraak, J.C. Influence of thermal conditions on the efficiency of high-performance liquid chromatographic columns, J. Chromatogr., 1983, 282, 399-412.
14. Mayr, G.; Welsch, T. Influence of viscous heat dissipation on efficiency in high-speed high-performance liquid chromatography, J. Chromatogr. A, 1999, 845, 155-163.
15. Poppe, H.; Kraak, J.C.; Huber, J.F.K.; van den Berg, J.H.M. Temperature gradients in HPLC columns due to viscous heat dissipation, Chromatographia, 1981, 14, 515-523.
16. Gritti, F.; Guiochon, G. Measurement of the axial and radial temperature profiles of a chromatographic column: Influence of thermal insulation on column efficiency, J. Chromatogr. A, 2007, 1138, 141-157.
17. Guiochon, G. The limits of the separation power of unidimensional column liquid chromatography, J. Chromatogr. A, 2006, 1126, 6-49.
18. van Deemter, J.J.; Zuiderweg, F.J.; Klinkenberg, A. Longitudinal diffusion and resistance to mass transfer as causes of nonideality in chromatography, Chem. Eng. Sci., 1956, 5, 271-289.
19. Giddings, J.C. Dynamics of Chromatography, Part I. Principles and Theory, Marcel Dekker, New York, 1965.
20. Grushke, E.; Snyder, L.R.; Knox, J.H. Advances in Band Spreading Theories, J. Chromatogr. Sci., 1975, 13, 25-37.
21. Mutton, I.M. Use of short columns and high flow rates for rapid gradient reversed-phase chromatography, Chromatographia, 1998, 47, 291-298.
22. Giddings, J.C. Comparison of theoretical limit of separating speed in gas and liquid chromatography., Anal. Chem., 1965, 37, 60-63.
23. Knox, J.H. Practical Aspects of LC Theory, J. Chromatogr. Sci., 1977, 15, 352-364.
24. Poppe, H. Some reflections on speed and efficiency of modern chromatographic methods, J. Chromatogr. A, 1997, 778, 3-21.
25. MacNair, J.E.; Lewis, K.C.; Jorgenson, J.W. Ultrahigh-Pressure Reversed-Phase Liquid Chromatography in Packed Capillary Columns, Anal. Chem., 1997, 69, 983-989.
26. Thompson, J.W.; Mellors, J.S.; Eschelbach, J.W.; Jorgenson , J.W. Recent advances in ultrahigh-pressure liquid chromatography, LC-GC NA., 2006, 24, 16-22.
27. Jerkovich, A.D.; Mellors, J.S.; Jorgenson, J.W. The use of micrometer-sized particles in ultrahigh pressure liquid chromatography, LC-GC AP., 2003, 6, 8-12.
28. Wu, N.; Clausen, A.M. Fundamental and practical aspects of ultrahigh pressure liquid chromatography for fast separations, J. Sep. Sci., 2007, 30, 1167-1173.
29. Blue, L.E., Franklin, E.G.; Godinho, J.M.; Grinias J.P.; Grinias, K.M. Recent advances in capillary ultrahigh pressure liquid chromatography, J. Chromatogr. A, 2017, 1523, 17-39.
30. Zhu, C.; Goodall, D.M.; Wren, S.A.C. Elevated temperatures in HPLC: Principles and Applications to Small Molecules and Biomolecules, LC-GC AP., 2005, 8, 48-59.
31. Yan, B.; Zhao, J.; Brown, J.S.; Blackwell, J.; Carr, P.W. High-Temperature Ultrafast Liquid Chromatography, Anal. Chem., 2000, 72, 1253-1262.
32. Heinisch, S.; Rocca, J.L. Sense and Nonsense of high-temperature liquid chromatography, J. Chromatogr. A, 2009, 1216, 642-658.
33. Sheng, G.; Shen, Y.; Lee, M.L. Elevated temperature liquid chromatography using reversed-phase packed capillary columns, J. Micro. Sep., 1997, 9, 63-72.
34. Vanhoenacker, G.; Sandra, P. High temperature and temperature programmed HPLC: possibilities and limitations, Anal. Bioanal. Chem., 2008, 390, 245-248.
35. Vanhoenacker, G.; Sandra, P. Elevated temperature and temperature programming in conventional liquid chromatography – fundamentals and applications, J. Sep. Sci., 2006, 29, 1822-1835.
36. McNeff, C.V.; Yan, B.; Stoll, D. R.; Henry, R.A. Practice and theory of high temperature liquid chromatography, J. Sep. Sci., 2007, 30, 1672-1685.
37. Greibrokk, T.; Andersen, T. High-temperature liquid chromatography, J. Chromatogr. A, 2003, 1000, 743-755.
38. Kirkland, J.J. Superficially porous silica microspheres for the fast high-performance liquid chromatography of macromolecules, Anal. Chem., 1992, 64, 1239-1245.
39. DeStefano, J.J.; Langlois, T.J.; Kirkland, J.J. Characteristics of superficially-porous particles for fast HPLC: some performance comparisons with sub-2-µm particles, J. Chromatogr. Sci., 2008, 46, 254-260.
40. Kaczmarski, K.; Guiochon, G. Modelling of the mass-transfer kinetics in chromatographic columns packed with shell and pellicular particles, Anal. Chem., 2007, 79, 4648-4656.
41. Fekete, S.; Fekete, J.; Ganzler, K. Shell and small particles: evaluation of new column technology, J. Pharm. Biomed. Anal., 2009, 49, 64-71.
42. Hayes, R.; Ahmed, A.; Edge, T.; Zhang, H. Core-shell particles: preparation, fundamentals and applications in high performance liquid chromatography, J. Chromatogr. A, 2014, 1357, 36-52.
43. Gritti F.; Guiochon, G. Facts and legends about columns packed with sub-3-µm core-shell particles, LC-GC NA., 2012, 30, 586-595.
44. Ruiz, V.G.; Olives, A.I.; Martin, M.A. Core-shell particles lead the way to renewing high-performance liquid chromatography, Trends Anal. Chem., 2015, 64, 17-28.
45. Cabooter, D.; Broeckhoven, K.; Sterken, R.; Vanmessen, A.; Vandendael, I.; Nakanishi, K.; Deridder, S.; Desmet, G. Detailed characterization of the kinetic performance of first and second generation silica monolithic columns for reversed-phase chromatography separations, J. Chromatogr. A, 2014, 1325, 72-82.
46. Sklenarova, H.; Chocholous, P.; Koblova, P.; Zahalka, L.; Satinsky, D.; Matysova, L.; Solich, P. High-resolution monolithic columns – a new tool for effective and quick separation, Anal. Bioanal. Chem., 2013, 405, 2255-2263.
47. Hormann, K.; Mullner, T.; Bruns, S.; Holtzel, A.; Tallarek, U. Morphology and separation efficiency of a new generation of analytical silica monoliths, J. Chromatogr. A, 2012, 1222, 46-58.
48. Guiochon, G. Monolithic columns in high-performance liquid chromatography, J. Chromatogr. A, 2007, 1168, 101-168.
49. Deeb, S. E.; Schepers, U.; Watzig, H. Fast HPLC method for the determination of glimepiride, glibenclamide, and related substances using monolithic column and flow program, J. Sep. Sci. 2006, 29, 1571-1577.
50. Badman, E. R.; Beardsley, R. L.; Liang, Z.; Bansal, S. Accelerating high quality bioanalytical LC/MS/MS assays using fused-core columns, J. Chromatogr. A, 2010, 878, 2307-2313.
51. Shaaban, H.; Gorecki, T. Fused core particles as an alternative to fully porous sub-2-µm particles in pharmaceutical analysis using coupled columns at elevated temperatures, Anal. Methods, 2012, 4, 2735-2743.
52. Ahmad, I.A.H.; Soliven A.; Allen, R.C.; Filgueira, M.; Carr, P.W. Comparison of core-shell particles and sub-2µm fully porous particles for use as ultrafast second dimension columns in two-dimensional liquid chromatography, J. Chromatogr. A, 2015, 1386, 31-38.
53. McCalley, D. Instrumental Considerations for the effective operation of short, highly efficient fused-core columns. Investigation of performance at high flow rates and elevated temperatures, J. Chromatogr. A, 2010, 1217, 4561-4567.
54. Schuster, S.A.; Boyes, B.E.; Wagner, B.M.; Kirkland, J.J. Fast high performance liquid chromatography separations for proteomic applications using fused-core silica particles, J. Chromatogr. A, 2012, 1228, 232-241.
55. Rogeberg, M.; Wilson, S.R.; Malerod, H.; Lundanes, E.; Tanaka, N.; Greibrokk, T. High efficiency, high temperature separations on silica based monolithic columns, J. Chromatogr. A, 2011, 1218, 7281-7288.
56. Wu, J.T.; Zheng, H.; Deng, Y.; Unger, S.E. High speed liquid chromatography/tandem mass spectrometry using a monolithic column for high throughput bioanalysis, Rapid Commun. Mass Spectrom., 2001, 15, 1113-1119.
57. Deng, Y.; Wu, J.T.; Lloyd, T.L.; Chi, C.L.; Olah, T.V.; Unger, S.E. High speed gradient parallel liquid chromatography/tandem mass spectrometry with fully automated sample preparation for bioanalysis: 30 seconds per sample from plasma, Rapid Commun. Mass Spectrom., 2012, 16, 1116-1123.
58. Deng, Y., Zeng, H., Unger, S.E., Wu, J.T. Multiple-sprayer tandem mass spectrometry with parallel high flow extraction and parallel separation for high-throughput quantitation in biological fluids, Rapid Commun. Mass Spectrom., 2001, 15, 1634-1640.
59. Gonzales-Ruiz, V.; Olives, A.I.; Martin, M.A. Challenging core-shell stationary phases with the separation of closely related anti-cancer compounds: performance studies and application to drug quantitation in cell cultures with multi-well plate clean-up, J. Chromatogr. A, 2014, 1364, 83-95.
60. Sander, L.C.; Pursch, M.; Wise, S.A. Shape selectivity for constrained solutes in reversed-phase liquid chromatography, Anal. Chem. 1999, 71, 4821-4830.
61. Sander, L.C.; Wise, S.A. Subambient temperature modification of selectivity in reversed-phase liquid chromatography, Anal. Chem. 1989, 61, 1749-1754.
62. Sander, L.C.; Wise, S.A. Shape selectivity in reversed-phase liquid chromatography for the separation of planar and non-planar solutes, J. Chromatogr. A, 1993, 656, 335-351.
63. Rimmer, C.A.; Lippa, K.A.; Sander, L.C. Shape Selectivity in Reversed-Phase Liquid Chromatography, LC-GC NA., 2008, 26, 984-998.
64. Lippa, K.A.; Rimmer, C.A.; Sander, L.C. editors, Shape Selectivity in Reversed-Phase Liquid Chromatography, Advances in Chromatography, CRC Press, 2007.
65. Shollenberger, D.; Cramer, H.; Bell, D.S. Evaluation of retention and selectivity using biphenyl stationary phases, LC-GC NA., 2017, 35, 360-365.
66. Zhou, S. N.; Reiner, E.J.; Marvin, C.H.; Helm, P.A.; Shen, L.; Brindle, I.D. Liquid chromatography/atmospheric pressure photoionization tandem mass spectrometry for analysis of dechloranes, Rapid Commun. Mass Spectrom., 2011, 25, 436-442.
67. Powell, M.; D’Arcy, M.B. Liquid phase separation of structurally-similar steroids using phenyl stationary phases, Anal. Methods, 2013, 5, 5014-5018.
68. Young, J.E.; Matyska, M.T.; Azad, A.K.; Yoc, S.E.; Pesek, J.J. Separation differences among phenyl hydride, UDC cholesterol and bidentate C8 stationary phases for stability indicating methods of tetracyclines, J. Liq. Chromatogr. Relat. Technol., 2013, 36,
926-942.
69. Pesek, J.J.; Matyska, M.T.; Dawson, G. B.; Wilsdorf, A.; Marc, P.; Padki, M. Cholesterol bonded phase as a separation medium in liquid chromatography: Evaluation of properties and applications, J. Chromatogr. A, 2003, 986, 253-262.
70. Bocian, S.; Matyska, M.; Pesek, J.; Buszewski, B. Study of the retention and selectivity of cholesterol bonded phases with different linkage spacers, J. Chromatogr. A, 2010, 1217, 6891-6897.
71. Bocian, S.; Soukup, J.; Matyska, M.; Pesek, J.; Jandera, P.; Buszewski, B. The influence of the organic modifier in hydro-organic mobile phase on separation selectivity of steroid hormones separation using cholesterol-bonded stationary phases, J. Chromatogr. A, 2012, 1245, 90-97.
72. Horvath, C.G.; Lipsky, S.R. Peak capacity in chromatography, Anal. Chem., 1967, 39, 1893-1893.
73. Bristow, P.A.; Knox, J.H. Standardization of test conditions for high performance liquid chromatography columns, Chromatographia, 1977, 10, 279-289.
74. Wang, X.; Barber, W.E.; Carr, P.W. A practical approach to maximizing peak capacity by using long columns packed with pellicular stationary phases for proteomic research J. Chromatogr. A, 2006, 1107, 139-151.