Supercritical fluid (SFC), green chromatography
Published over 11 years ago. See the latest and most current information on Supercritical fluid (SFC), green chromatography.
As the old saying goes, “Perception is reality” and for many scientists, the acronym SFC, derived from the term supercritical fluid chromatography, carries with it many unfavourable connotations that might have impeded the wide adoption of this technique within the modern laboratory. This may have something to do with the unreliable SFC systems of yesteryear. It could be also due to some unrealistic promises made by early SFC proponents. It may also have just as much to do with the vocabulary of SFC. This article will explain why the word “supercritical” is misleading, why it should be retired and then go on to explain what scientists should know about modern SFC that may begin to dispel the mystique around it and erase the negativity. It may also shed some light for readers on why there is a resurgence of interest in the technique among analytical scientists for both chiral and achiral chromatography.
Origins of Supercritical Fluid Chromatography
Supercritical fluid chromatography was invented by gas chromatographers [1] exploring gases under high pressure hoping to elute compounds that could not be analysed with GC because the analytes are prone to decompose at the temperatures needed to elute them. To compensate, Klesper et al. [1] used higher pressures to elute porphyrin mixtures and therefore reduce the higher temperature needed for solute elution. Supercritical temperatures were maintained to enable the gas pressure to be continuously increased without it passing through the vapor-liquid biphasic conditions. They used dichlorodifluoromethane (Tc = 112°C) and monochlorodifuloromethane (Tc = 96°C) at the pressures above 1000 psia and 1400 psia, respectively.
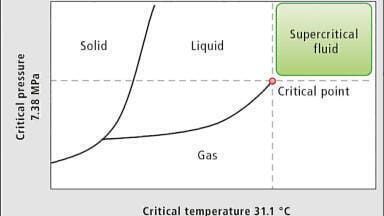
Carbon dioxide (CO2), which is the main solvent used in SFC systems today, was first used by Sie et al. [2]. Many other solvents, for example ammonia (NH3) and nitrous oxide (N2O) were tested for use with SFC but CO2 prevailed as a principal mobile phase because of several advantageous properties [3] (e.g. non-toxicity, non-flammability) and the critical point (Tc=31°C, Pc=1070 psia) being closer to standard ambient temperature and pressure.
Working within Supercritical Conditions
What many scientists may not realise is that moving from liquid or gaseous state to a supercritical condition does not bring any sudden change in physical properties to a solvent, at least not anything that will influence chromatographic performance. The main difference between solvents in their liquid state and solvents in a supercritical condition is their compressibility, which gradually increases from liquid to supercritical conditions. Higher compressibility makes solvents “tunable”, which means that by varying the pressure and/or the temperature of the mobile phase inside the column one can greatly manipulate chromatographic performance without adding any organic modifiers. This feature of SFC, coupled with the use of “carbon-neutral” CO2 as the principal solvent, makes the technology “green” and has attracted many applications.
But working with compressible fluids has a price. Almost all the problems related to early instrument design originated from the compressibility effects. Pumping highly compressible fluids and maintaining a constant pressure inside the system can be very challenging. Additionally, both the pump and the automated back-pressure regulator or ABPR (the control valve which maintains the system pressure) generate pressure ripples which create detector noise. Fortunately most of these instrumental challenges have now been solved and the currently available instruments are as robust as HPLC or UPLC/UHPLC instruments.
Early Misconceptions
Due to the inability to clearly understand the foremost advantages of working with CO2, the early proponents of SFC made some unrealistic claims. The predominant catchphrase then - ‘liquid-like dissolving power and gas-like viscosity’ - over-simplified reality. These are contradictory conditions in that there is a trade-off between these two properties across the supercritical space. Additionally, it was determined [4] that with neat CO2, in the regions where viscosity is gas-like, even small pressure drops lead to expansion cooling which significantly affects the efficiency.
More importantly, the dissolving power of supercritical CO2 was not as good as it was initially estimated [5] specifically for relatively polar analytes. To expand the range of applications, researchers started mixing organic modifiers such as methanol, ethanol, and acetonitrile with the CO2. This practice raised a question about the supercritical condition of the solvent mixtures. If the use of organic modifiers raises the critical temperature or pressure of the mixture, how do practitioners know if the mobile phase is now super- or subcritical? Or does it matter? Researchers who started working at temperatures that are now clearly below the supercritical temperature of the mixture, even below the Tc of CO2 (31°C), began to realise they could generate excellent chromatography. What practically matters is to avoid the boiling of the mobile phase in the column and the detector, for understandable reasons [6]. This led to the practice of employing so-called subcritical fluid chromatography as a re-defined use of SFC, complicating for many the understanding of what SFC is and isn’t.
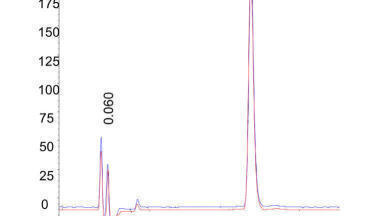
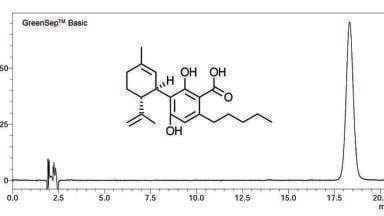
To resolve this debate, we performed a set of experiments to run across the critical isotherm at various randomly selected pressure-temperature conditions designed to show chromatograms for both subcritical and supercritical conditions (Figure 1). Neat CO2 was used for all the experiments. It can be noted from the chromatograms that unless they were clearly marked it is not possible to identify the chromatograms which were obtained at supercritical conditions and those at subcritical conditions. In other words, Figure 1 demonstrates that supercritical conditions do not render any special property to the chromatography and does not have to be a precondition for separation using CO2 as a mobile phase, calling into question the need for a debate in the first place.
CO2 as a Solvent
If supercritical CO2 is not a precondition for SFC then the term “supercritical fluid chromatography” is outdated and a misnomer. And if that is the case, what should we consider based on current practices?
At lower temperatures, technically at liquid conditions, the CO2-based mobile phase is not as strongly tunable and the separation is less susceptible to pressure variation. Today’s practitioners focus on using different organic modifiers, wider % solvent gradients (both <5% and >45%), and more mass spectrometry-friendly additives to achieve successful separations at these lower temperatures. For example at 2,000 psia and 20°C the compressibility of a CO2 + methanol (70/30, mol/mol,%) mixture is close to that of methanol, but the viscosity is still more than three times less (ref. Table 1). This reduced viscosity can be exploited for sharper, faster gradient profiles.
So, if the supercritical condition does not offer any special properties and we gradually move away from the original tunable conditions to the newer more robust conditions, what is left in the SFC descriptor? The answer is CO2 - and thinking of liquid CO2 as a main co-solvent in chromatography. In practice, SFC systems today are so similar to HPLC or UPLC/UHPLC systems in terms of the way they modulate mobile phase properties with organic modifiers and additives, that many scientists familiar with HPLC have told us that to learn to operate a present-day SFC system, such as the Waters® ACQUITY® UPC2®System, should take someone no more than 10 minutes.
Breaking the Barriers -
Converging Methods
The advantage of a CO2-based mobile phase is that it is compatible with a multiple range of stationary phases and its wide miscibility range with polar organic modifiers. Why is that important? Currently there are several modes of HPLC – e.g normal-phase chromatography, reversed-phase chromatography etc. The polarity of the analytes determines the choice of the stationary phase chemistries as well as the mobile phases and, very often, the systems and expertise in the laboratories to work with them.
The primary workhorse of analytical HPLC is reversed-phase chromatography which separates analytes mainly based on non-polar interactions. The stationary phase for reversed-phase is non-polar (e.g. C18 bonded silica) and the main solvent in the mobile phase is highly polar - most typically water. Retention of analytes are controlled by mixing in a less polar modifier, (e.g. acetonitrile, methanol) as a function of time imparting a solvent gradient. Using a C18 column and with the properly chosen combination of modifiers, gradients and additives, one can achieve a successful separation. However for extremely polar or extremely non-polar analytes or for chiral separation, reversed-phase is not the mode of choice and we change to normal-phase chromatography.
In normal-phase the stationary phase is polar and we use non-polar primary solvents such as heptane or hexane along with a small percentage of relatively polar modifiers (e.g. ethyl acetate, methylene chloride, ethylene chloride, IPA and ethanol). The elution order of compounds is opposite that of reversed phase chromatography, with non-polar compounds eluting earlier than polar compounds that are more retained. It is the ability to deal with both polar and non-polar compounds that gives normal phase its appeal. Normal-phase is also used predominately in chiral separations due to the polar nature of chiral stationary phases. Reversed phase chromatography does not have such an attractive range of suitable phases for chiral work.
There are, however, several challenges associated with normal phase chromatography including;
•
the difficulty of doing solvent gradients due to miscibility issues, which makes the method development and analysis times longer.
•
the homogeneity of the silica surface and possible effects that small amounts of water / polar mobile phase can have on the retention time (CO2-based mobile phases are much more robust with respect to retention time reproducibility)
•
the solubility of polar compounds in the mobile phase
•
potential worker exposure and environmental effects from the use of chlorinated solvents
•
inability to use low UV wavelengths with many modifier solvents
Matching correctly, the diverse isocratic non-polar solvent mixtures with corresponding stationary phases, is difficult and that is why normal phase and chiral analysis became somewhat specialised techniques separated from the mainstream.
Modern SFC is breaking down these barriers. The property of CO2 (i.e. its miscibility) with a wide range of polar organic solvents has made the CO2-based mobile phase versatile enough to separate compounds of a very wide polarity range. Not only can we use CO2-based solvents with both polar and non-polar stationary phases (including those originally used for reversed phase), we can influence the chromatography by modulating solvent gradients with both the phases, leading to a much wider choice of columns and separation methods.
So the unique feature of modern SFC is not the physical state or the condition of the solvent, but rather the ability to combine or converge the separation of a much wider variety of compounds with one system. Chemists do not need to be trained in normal-phase and reversed-phase chromatography separately. Its acceptance for standard workflows in analytical laboratories is also more streamlined with the simple addition of a standard CO2 cylinder.
Conclusion
To summarise, it is time scientists see SFC in a new light: modern SFC is like an LC separation technique that happens to use liquid CO2 as its primary mobile phase. The tunability of supercritical CO2 as a single solvent without organic co-solvents may be useful for niche application areas but it is not crucial for many of today’s separations or analysis.
CO2-based solvent mixtures, together with today’s column chemistries, work fantastically well, and bring a convergence between both chiral and achiral separations, as well as reversed-phase and normal-phase chromatography. When mixed with a range of polar organic co-solvents a single CO2-based analytical system can perform separations that often require multiple dedicated systems in a laboratory using widely different mixtures of solvents and expertise to work with them.
This article attempted to explain why we should move out from a state-specific name to an application-specific name, or why we started calling modern SFC - Convergence Chromatography - which may help in making the technique more readily acceptable to future users.
References
[1]
K. Klesper, A.H. Corwin and D.A. Turner, J. Org. Chem., 27, 700 (1962)
[2]
S.T. Sie, W. Van Beersum, and G.W.A. Rjinders, Sep. Sci., 1(4), 459 (1966)
[3]
G. Guiochon and A. Tarafder, Journal of Chromatography A, 1218 (2011) 1037–1114
[4]
D. P. Poe, J. J. Schroden, J. Chromatogr. A 1216 (2009) 7915-7926.
[5]
T. A. Berger, Packed Column SFC, RSC Chromatography Monographs, 1995
[6]
P. Sandra, B. Szucs, R. Sandwich, Hanna-Brown, in: HPLC-2009, 2009. pp.
Communication L-1.
[7]
E.W. Lemmon, M. Huber, M.O. McLinden, D.G. Friend, National Institute of Standards and Technology, Standard Reference Data Program, Gaithersburg 9.1.and Technology, Standard Reference Data Program, Gaithersburg 9.1.