Supercritical fluid (SFC), green chromatography
Published over 6 years ago. See the latest and most current information on Supercritical fluid (SFC), green chromatography.
Some manufacturers of current supercritical fluid chromatographs (SFC’s) routinely claim that their products are ‘ultra-high performance’, similar to ultra-high performance liquid chromatographs (UPLC’s - here defined as reduced plate height ≈ 2 with sub-2 µm particles in 2.1 mm internal diameter (ID) columns). However, none of the current commercially plumbed SFC instruments are any better in terms of extra-column dispersion than instruments available in 1992, which were specifically designed for use with 4.6 mm ID, 150-250 mm long columns packed with 5 µm particles. Despite the fact that fast and ultra-fast separations are an important recent development, today, most users appear to be uninterested or unwilling to make any of the modifications necessary to approach true ultra-high performance SFC.
It is often stated that column technology has outstripped instrumentation, but this does not mean columns 3 or 2.1 mm ID are all well packed. A few groups have attempted to modify commercial SFC instruments to try to determine the quality of small diameter columns packed with sub-2 µm particles. In fact, the results from these studies suggest that few columns used in these reports were actually well packed. The results were often confusing and counter-intuitive, due to the convolution of poor packing and excessive extra-column effects. Thus, both the instruments and the columns were inadequate. This review describes the various attempts, the often confusing results, and a path forward.
The use of 2.1 mm ID columns, packed with sub-2 µm particles, producing hmin ≈ 2, defines ultra-high performance liquid chromatography (UHPLC). In order to achieve a minimum reduced plate height (hmin) approaching 2, on such columns, very low extra-column dispersion, on the order of a few µL2, or smaller, is required. Compared to conventional HPLC’s, UHPLC’s require shorter lengths of 100 or 125 µm vs. 175 µm connector tubing, smaller detector flow cells, often less than 1 µL, and smaller injection volumes. In UHPLC such small particles require very high pressure pumps, capable of 1000 bar or more. In UHPLC, the very large pressure drops required result in an increase in the mobile phase temperature [1-3], due to the expansion of the mobile phase. Small ID columns, such as 2.1 or even 1 mm, are thought necessary to minimise the length of radial thermal gradients, caused by the decompression of the mobile phase. Radial gradients potentially cause serious loss of efficiency. Smaller ID columns require smaller optimum flow rates.
Supercritical fluid chromatography (SFC) is inherently 3 to 5 times faster than HPLC on the same sized columns, due to lower viscosity [4] and higher diffusivity of/in the mobile phase. Pressure drops are 1/3rd to 1/5th those encountered in HPLC. Thus, pressure drops are usually no more than a few hundred bar. Since the pressure drops in SFC are much lower, high pump pressure capability is much less important, compared to UHPLC.
Modified CO2 mixtures can cool, warm or maintain the same temperature when expanded [5], depending on modifier concentration. The combination of low pressure drops and modest levels of warming or cooling seldom, if ever, distort peaks or cause losses of efficiency. Thus, the potential for forming significant radial thermal gradients in SFC is much lower than in UHPLC. One would think that the increasing use of sub-2 µm particles in UHPLC, with the associated large pressure drops, and the resulting large thermal gradients should stimulate the development of ultra-high performance supercritical fluid chromatography (UHPSFC), since in SFC those issues are greatly reduced. However, there have been no attempts by the vendors of such equipment toward developing true UHPSFC.
Minimising thermal gradients has been the main justification for the use of 2.1 mm columns in UHPLC, but since such gradients are much less likely in SFC, the need for such columns has less justification. Columns 3 mm ID, with the same sub-2 µm packings, have 4X the dispersion compared to 2.1 mm columns, allowing 4X higher extra column dispersion in the system. It appears that 3 mm ID columns are, at present, easier to pack, and more efficient, compared to 2.1 mm columns. Further, a 2.1 mm ID column can tolerate only ≈ 1/2 the injection volume of a 3 mm ID column. Never the less, the future development of SFC instrumentation ought to aim for the use of sub-2 µm particles in 2.1 µm ID columns with minimal extra-column dispersion (hmin ≈ 2), even though there is little current compelling justification for such development.
It appears to be a common perception that columns are inherently well packed, supported by the fact that column technology has largely outstripped instrumentation. It is clear that the SFC instrument configurations shipped from the manufacturers are inadequate for use with sub-2 µm particles. However, many of the modifications used to improve HPLC instruments to UHPLC performance, such as shorter, smaller ID tubing, and smaller detector cell volumes have resulted in confusing and anti-intuitive results when used in SFC. A major problem is that it is unclear, a priori, how well columns are actually packed, and whether any poor efficiency observed is due to the column, the instrumentation, or both.
There have been a modest number of attempts to characterise the improvements needed to achieve the goal of hmin ≈ 2 with sub-2 µm particles. This report summarises those attempts.
In 2012, Guillarme [6] compared a Waters UHPLC to a Waters UPC2 which he called a UHPSFC. Both chromatographs were used as plumbed by the manufacturer. The UHPLC was found to have an extra-column variance of ≈ 3 µL2. The tubing between the injection valve and the column was 250 mm long, 130 µm ID, including a passive heater. The tube between the column and detector was 150 mm long, 100 µm ID. The injector had a 5 µL loop. The detector flow cell was 0.5 µl.
On the other hand, the SFC had a nearly 30X higher variance of ≈ 85 µL2, with 600 mm of 175 µm tubing before the column and 600 µL of 175 µm tubing after the column. The injector had a 10 µL loop. The flow cell was 8 µL. This very high extra-column dispersion in the SFC negates any pretence that the results could be called ‘ultra-high performance’. However, most other current commercial SFC’s have similar, very high extra-column dispersion [7] as plumbed and shipped by their manufacturers, and are similar to conventional HPLC’s and SFC’s in the 1990’s.
It is informative to calculate the theoretical dispersion of various column sizes in order to estimate the extra-column dispersion allowed. Assuming a particle size of 1.8 µm, the optimum flow rate on various ID columns can be estimated from experience at ≈ 2 mL-min-1 for 3 mm ID columns and 1 mL-min-1 in 2.1 mm columns. Knowing column length allows calculation of retention times and peak widths. At k = 2, h = 2, the dispersion of a 3x100 mm column is ≈ 73 µL2, whereas a 2.1x100 mm column has a dispersion of only ≈ 17.5 µL2. A rough ‘rule of thumb’ says that extra-column dispersion should be < 1/5th column dispersion. This suggests that the extra-column dispersion should be < 3.5 µL2 when using a 2.1x100 mm column with 1.8 µm particles.
Guillarme stated that although 2.1 mm columns tend to be favoured in UHPLC, they could not be used in SFC due to the high extra-column dispersion of the commercial systems. He proceeded to compare the performance of the 2 instruments using 2.1 mm ID reversed phase columns in UHPLC, but 3 mm ID normal phase columns in UHPSFC, all with 1.7 µm packings. The SFC was found to be 4 times faster, but had a minimum reduced plate height (hmin) > 2.8, while the UHPLC had hmin ≈ 2.2. The 3x100 mm column should have 4X the column dispersion, allowing 4X higher extra-column dispersion, compared to the 2.1 mm column, but still exhibited substantially worse efficiency. Guillarme made no attempt at improving the performance of the SFC, through changing the tubing or UV flow cell, but he clearly outlined the major differences between the UHPLC and the SFC.
On the other hand, Broeckhoven stated [8] that 2.1 µm ID columns must be used in SFC in order to achieve high velocities with commercial instruments, due to the high optimum flow rates of the columns and limited maximum flow rates of the pumps. However, he also agreed that the extra-column dispersion was excessive, as shipped by the manufacturer. Much of the extra-column dispersion occurs in the connector tubing and to a lesser extent in the detector flow cell. The instrument used was an Agilent 1260. Attempting to minimise such extra-column dispersion, he varied the composition of the sample solvent, the injection volume, and the size and length of the pre- and post-column tubing, and detector volume.
Tubing ID in front of the column was decreased from 175 µm to 125 µm, and the shorter (125 mm), built-in heat exchanger (HX) was used (with 175 µm internal tubing). The standard 13 µL detector flow cell was replaced with a 1.7 µL cell, with a 310 mm long, 125 µm inlet tube, and eventually with a 0.6 µL flow cell.
There were further tests where a 15 cm length of 65, 120, 170, and 250 µm tubes were inserted between the column outlet and the detector inlet, with no significant effect.
The plumbing modifications made were similar to those made to create the early UHPLC’s, and should have decreased extra-column variance by more than an order of magnitude. Surprisingly, these plumbing changes had minimal effect on total dispersion using either a 2.1x150 mm, HILIC, or a 2.1x100 mm HILIC 1.8 µm column. With a little retained solute, k = 2.3, the reduced plate height was h ≈ 4, and a long retained solute, k = 10.8, h ≈ 2.87. The latter is similar to Guillarmes [6-7] results with an unmodified SFC, with a extra-column variance of ≈ 80 µL2 using a 3 mm ID column.
Aspects of the injection volume and the sample solvent composition were found to be most important in determining efficiency. The fixed injection loop was replaced with a length of 50 µm ID tubing yielding a fixed injection volume of 0.4 µL. The sample solvent consisted of a 1:1 mixture of hexane/IPA with various concentrations of ethanol added. Above 10% ethanol there was noticeable loss of efficiency. This superficially appears to be a ‘strong sample solvent’ effect although the sample isn’t really strong. The mobile phase was 8% methanol in CO2 which is probably not a much different solvent compared to 10% ethanol in hexane:IPA. The results all suggest that the columns were probably relatively poorly packed, but the characteristics of the injection problems were difficult to explain.
In 2015 while developing ultra-fast chiral SFC separations, Armstrong [11] stated: “...column technology is ahead of existing chromatographic instruments”. He meant, of course, SFC instruments, as delivered by the manufacturers, were inadequate to see the full efficiency of columns packed with sub-2 µm particles. A Jasco semi-prep SFC was used with 4.6x50 mm column packed with 1.9 µm teicoplanin particles. The optimum flow on such a 4.6 mm ID column is ≈ 3.5-5 mL-min-1, depending on modifier concentration. In an effort to minimise extra-column effects, near optimum linear velocity, various ID tubes; 50, 75, 127, 254, and 508 µm, of PEEK or silica lined PEEK were used, with 11.5 cm in front of and 20 cm after the column. The column oven was bypassed (no thermal control of column temperature). Rather surprisingly, it was found that 254 µm tubing marginally produced the highest plate counts. These results are also non-intuitive. However, the differences in efficiency between 75, 120, and 254 µm tubing were minimal (≈ 2%), and all were poor, with hmin ≈ 5.85, k < 2. The smallest tube ID ought to yield the lowest dispersion, but did not. The most reasonable conclusion was that this column was also not very efficient.
Many of the plumbing modifications made by Broeckhoven [8] were similar to those previously made by Berger [9] in 2010 when the first use of sub-2 µm particles in SFC was demonstrated. The chromatograph consisted of an Agilent 1260SL HPLC, with an Aurora SFC conversion module. A 10cm length of 125 µm tubing was used as the injection loop (≈ 1.25 µL). The sample solvent was methanol. All the tubing from the injection valve to the detector was 125 µm ID stainless steel tubing of the shortest possible length (≈70 cm total). However, the 2 heat exchangers with 175 µL tubing were retained. The photodiode array detector flow cell was the same 1.7 µL, 6 mm cell used by Broeckhoven [8]. A larger ID 3x100 mm column packed with 1.8 µm particles delivered a reduced plate height of ≈ 2.48 at k = 3.63, which was somewhat better than [8]. The same instrument was used to perform the first use of superficially porous 2.6 µm particles [10] in SFC. The Kinetex 2.6 µm column was 4.6x150 mm, and produced reduced plate heights as low as 1.62.
Later, Berger [12] modified a commercial Agilent 1260 Series I instrument (a more developed instrument compared to [9,10]) to achieve a reduced plate height of 2 with k = 2, using 3x100 mm columns packed with 1.8 µm particles. Many of the modifications were the same as used previously [8,9]. The injection loop was replaced with a 10 cm length of 125 µm tubing (≈ 1.25 µL). The sample solvent was methanol. The electronic filter was set to 80 Hz. All the 175 µm connector tubing was replaced with 125 µm versions, as before, but, in this case, the built-in heat exchangers (25 cm and 12.5 cm of 175 µm ID) were replaced with low dispersion HX’s (125 µm ID). The column was connected to the detector with 510 mm of 125 µm tubing (including the HX). The 13 µL detector flow cell was replaced with a 2 µL, 3 mm path length version. The measured variance (using w1/2) was ≈ 7 µL2.
Achieving h ≈ 2 required very well packed columns. In fact, a fairly large number (8) of 3x100 mm columns with 1.8 µm particles were tested before several columns were found to produce reduced plate heights of ≈ 2 at k =2. Without such highly efficient columns, there is always the question of what is real. Since 6 of 8 columns failed to produce reasonable efficiency, while the calculated plumbing dispersion showed it was feasible, the rest were poorly packed. Attempts to use 2.1x100 mm columns with 1.8 µm particles yielded poor efficiencies (hmin = 2.85) even with k > 8 [12]
The same instrument and modifications were used to produce a hmin = 1.93 with a 4.6x50 mm, 1.8 µm R,R Whelk-O chiral column [13]. This column had a theoretical column variance of ≈ 220 µL2, at k = 2. The plumbing was adequate for use with 1.8 µm particles in such a column, since an extra-column dispersion of only < 44 µL2 was required. On the other hand, h = 2.78 was obtained using a 3x50 mm, 1.6 µm column packed with an immobilised polysaccharide (IA-U) chiral column [14], using the same plumbing (column dispersion ≈ 36.8 µL2). Since the extra-column dispersion was ≈ 1/5th the column dispersion it probably indicates the column was relatively poorly packed or the chiral stationary phase was heterogeneous.
Gasparrini [15] modified a Waters UPC2 SFC for use with 1.8 µm particles with another chiral selector (Whelko-O1). He replaced the 600 cm long, 175 µm ID tubes in front of and after the column with 250 mm, and 350 mm tubes respectively. The ID of these tubes was progressively decreased from 180, to 130, to 100 µm. This shortening of the tubing apparently involved the oven design which was replaced with an in-house built unit. Similarly, the autosampler with a 10 µL loop, was replaced with an external valve with a fixed 200 nL loop. This external, very small loop injector seemed to eliminate (or avoid) the injection problems of Broeckhoven [8]. He also replaced the 8 µL detector flow cell with a 3 µL cell. With all the modifications in place he obtained σ2e-c ≈ 2.2 µL2, producing h = 1.88 on a 4.6x50 mm column with 1.8 µm R,R-Whelk-O1 particles. This is probably the lowest reduced plate height ever using a chiral stationary phase, let alone one with 1.8 µm particles.
Gasparrini paid particular attention to the onset of turbulent flow [16,17] and its beneficial effects on reduced extra-column dispersion. However, he did not recommend the use of 100 µm tubing since it performed no better than the 130 µm tubes, but exhibited substantially higher back pressure. This lower pressure drop is probably more important with a Waters SFC system where the maximum pump pressure is only 400 bar.
In several brief communications Berger [18,19] recently used an Agilent 1260 Series II (different instrument from earlier reports). This system has an unusual autosampler design. There is no injection ‘loop’. A 0.1 µL sample injection was pushed into the flowing mobile phase with a high pressure syringe. However, the content of a needle seat capillary (75 µm, 0.46 µL) filled with IPA preceded the sample and a small plug of IPA followed the sample. This possibly focuses the injection [20]. The column was 3x20 mm, packed with 1.8 µm RX-Sil. The injection valve was connected to the column with 385 mm of 125 µm tubing and 105 mm of 75 µm tubing (in an ‘ultra-low dispersion’ HX) in front of column, and 31 cm of 125 µm after the column without a HX, The same 2 µL flow cell was used. Efficiency was somewhat disappointing [18] with hmin = 2.4. The extra-column volume was ≈11 µL. The injection volume was extremely important. Increasing injection volume of the sample dissolved in methanol resulted in fairly rapid loss of efficiency.
In a follow-up study [19], the same column was used but the plumbing was changed. A 270 mm long stainless steel tube of 75 µm ID connected the injection valve directly to the column inlet without a heat exchanger. A special 2 µL, 3 mm path length flow cell with a 380 mm long, stainless steel 75 µm ID inlet tube was fabricated by Agilent R&D, Waldbronn, DE, and was connected directly to the column outlet. There was no thermal control. The extra-column volume was ≈ 5.75 µL. The hmin was a remarkable 1.65, (k = 3.2, n = 7).
The equation [21] for dispersion in tubing with laminar flow is:
σ2t = π r4 L F/24 D
The radius was 0.00375 cm. The length, L, was 65 cm. Flow, F = 0.0333 cm3/sec. The diffusion coefficient was estimated as 7x10-5 cm2/sec. The calculated dispersion of the tubing was ≈ 0.800 µL2. Any turbulence in the flow would decrease this value.
The detector flow cell nominally acts as a mixing chamber, with an equation:
σ2d =V2/12
It is sometimes suggested that the denominator should be smaller at 6 or even 4. Calculated dispersion of the detector cell was 0.333 µL2. Thus, calculations of the combination of tubing and detector dispersion was as low as 1.13 µL2 and unlikely to be > 2.
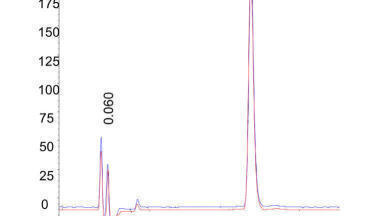
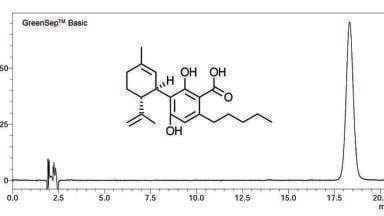
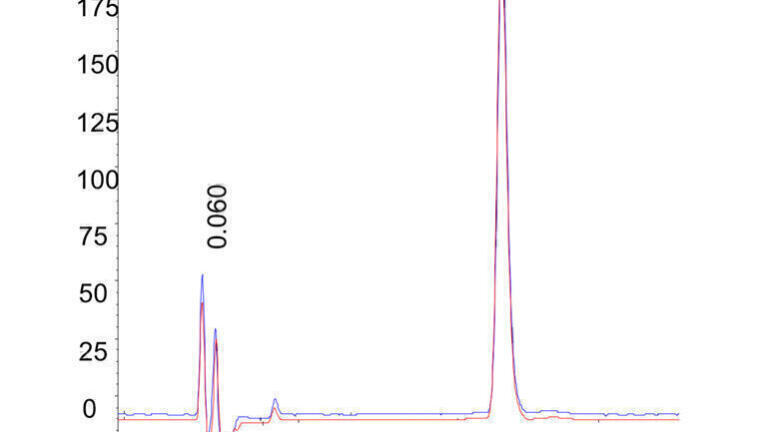
The column had a theoretical dispersion of ≈ 14.2 µL2 (h =2 @ k = 2). The observed total dispersion was 17.1 µL2 at k = 3.43. The high observed efficiency was quite surprising, particularly with such a short column and sub-2 µm particles. Two superimposed injections with an average h = 1.56 are presented in Figure 1.
The fastest peaks had w1/2 = 4.86e-3 min (292 ms)(120 Hz detector). The sample injection volume was 0.1 µL. The feed speed was 1000 µL/min or 16.7 µL/sec. Thus, the injection time was ≈ 60 ms.
The dispersion of the 3x20 mm column should compare favourably with the nominal column dispersion of a 2.1x100 mm column, packed with the same 1.8 µm particles, which is 17.7 µL2 (k = 2). However, attempts to use a number of 2.1 mm ID columns resulted in poor efficiency, even with 0.1 µL injections.
Although the use of 75 µm ID tubes have significant negative aspects, operating them near optimum flow rates on either 3 mm ID columns (≈2 mL-min-1) or on 2.1 mm ID columns (≈ 1 mL-min-1) mostly results in laminar flow in the tubing [17]. While turbulent flow is good for minimising dispersion in the tubing, it also results in major increases in extra-column pressure drops.
Many of the difficulties in obtaining high efficiency with small ID columns packed with sub-2 µm particles involve injection volume and injection solvent [22]. This is particularly true with smaller ID columns packed with sub-2 µm columns. In SFC one cannot dissolve the sample in the mobile phase, since it is essentially a compressed gas, that cannot be metered quantitatively into a vial, at least with current technology.
With larger columns (3-4.6 mm ID), the tendency in SFC, is to dissolve the sample in the modifier. However, this tends to make the ‘strong sample solvent effect’ a problem, by overwhelming the local mobile phase solvent strength on the column just after injection. It is particularly true when the injected sample volume is large, or at low modifier concentrations. Here, on small ID columns, ‘large’ can be 0.1 µL.
Decreasing the sample solvent polarity is desirable, as suggested by Broeckhoven [8], but many polar solutes are not soluble, or poorly soluble, in non-polar solvents like hexane/heptane. Adding IPA or EtOH obviously increases polarity/solubility but Guillarme [23] suggests that mixed sample solvents are always a problem and usually result in peak distortions. He also suggested a number of other pure aprotic sample solvents that should diminish the strong sample solvent effect, such as acetonitrile, methyl t-butyl ether (MTBE) and dichloromethane (DCM) as sample solvents but they do not seem very ‘green’.
Current commercial SFC’s are not plumbed by the manufacturers for use with sub-2 µm packings even though some characterise them as UHPSFC. However, it was demonstrated that true UHPSFC performance can be achieved with 3 mm and perhaps even 2.1 mm ID columns by minimising tubing length, tubing ID, and using smaller detector flow cells. It also required optimised injections in terms of sample solvent composition and injection volume. This ultimately requires columns that are actually well packed. The efficiency improvements in several of the works reported here are equivalent to at least the early UHPLC’s but with significantly faster speeds.
Claims that column technology has outstripped instrument development in SFC doesn’t necessarily mean that all columns are as efficient as expected. The lack of certainty in the inherent efficiency of the columns used in these reports has led to considerable confusion about the effect of tubing ID, detector flow cell effects, and injection issues. The modifications reported by most groups should logically have dramatically improved extra-column dispersion but in most cased didn’t appear to. It appears that most of the polar columns of 3 or 2.1 mm ID packed with sub-2 µm polar particles used in these studies were relatively poorly packed or exhibited mixed retention behaviour to the point where the column dispersion was large enough to make the extra-column dispersion fairly irrelevant .
Statements that column technology has outstripped instrumentation is only partly true with regard to sub-2 µm particles in 2.1 and 3 mm columns.
A major source of confusion appears to revolve around injection volume, selection of injection solvent, and the interactions between the sample, sample solvent, and their competition for sites on the stationary phase. On these small ID columns, efficient injections tend to be limited to very small volumes. While recent reports [22,23] have started to clarify some of these issues more work is needed.
The path forward seems fairly clear. Decreasing the extra-column variance by miniaturising tubing ID, length, and detector flow cell volumes is effective, but is not yet optimised. More effort is needed in optimising sample solvent characteristics to both the solutes and stationary phase. We need consistently more efficient columns of the appropriate dimensions. Fortunately, this seems to be getting better. It is hoped that the works summarised here, combined with future works, will eventually result in the instrument manufacturers producing a true UHPSFC.
1. Halasz, I., Endele, R., Asshauer, “Ultimate limits in high-pressure liquid chromatography” J., (1975) J. Chromatogr. 112, 37-60.
2. Horvath, C., Lin, H.J., “Band spreading in liquid chromatography: General plate height equation and a method for the evaluation of the individual plate height contributions”, (1978) J. Chromatogr. 149, 43-70.
3. de Villiers, A., Lauer, H., Szucs, R., Goodall, S., and. Sandra, P., Influence of frictional heating on temperature gradients in ultra-high-pressure liquid chromatography on 2.1 mm I.D. columns”, (2006), J. Chromatogr. A, 1113, 84-91.
4. Terry A. Berger, “Clarifying the Relationship Between Density and Viscosity of Methanol/Carbon Dioxide Mixtures used in Supercritical Fluid Chromatography”, Chromatography Today, Feb/March, 2019, pp. 28-31.
5. Sam O. Colgate, Terry A. Berger, “On axial temperature gradients due to large pressure drops in dense fluid chromatography”, 2015, J. Chromatogr. A, 1385, 94-102.
6. Perrenoud A.G-G., Veuthey, J-L. Guillarme, D., “Comparison of ultra-high performance supercritical fluid chromatography and ultra-high performance liquid chromatography for the analysis of pharmaceutical compounds”, 2012, J. Chromatogr. A, 1266, 158-167.
7. Alexandre Grand-Guillaume Perrenoud, Chris Hamman, Meenakshi Goel, Jean-Luc Veuthey, Davy Guillarme, Szabolcs Fetkete, “Maximizing kinetic performance in SFC using state-of-the-art instruments”, 2013, J. Chromatogr. A, 1314, 288-297.
8. Ruben De Pauw, Konstantin Shoykhet, Gert Desmet, Ken Broeckhoven, “Understanding and diminishing the extra-column band broadening in supercritical fluid chromatography”, 2015, J.Chromatogr. A, 1403, 132-137.
9. Terry A. Berger, “Demonstration of high speeds with low pressure drops using 1.8 µm Particles in Supercritical Fluid Chromatography (SFC)”, Chromatographia, 72 (2010) 596-602.
10. Terry A. Berger, “Characterization of a 2.6 μm Kinetex porous shell hydrophilic interaction liquid chromatography column in supercritical fluid chromatography with a comparison to 3μm totally porous silica”, 2011, J. Chromatogr. A, 1218, 4559-4568.
11. Chandran L. Barhate, M. Farooq Wahab, D.J. Tognarelli, Terry A Berger, Daniel W. Armstrong. “Instrumental Idiocyncracies Affecting the Performance of Ultrafast Chiral and Achiral Supercritical Fluid chromatography”, 2016, Anal. Chem., 88, 8664-8674.
12. Terry A. Berger, “Instrument modifications that produced reduced plate heights < 2 with sub-2 μm particles and 95% of theoretical efficiency at k = 2 in supercritical fluid chromatography”, 2016, J. Chromatogr. A, 1444, 129-144.
13. Terry A. Berger, “Kinetic performance of a 50 mm long 1.8 μm chiral column in supercritical fluid chromatography”, 2016, J. Chromatogr. A,1459, 136-144.
14. Terry A. Berger, “Preliminary kinetic evaluation of an immobilized polysaccharide sub-2 µm column using a low dispersion supercritical fluid chromatograph”, 2017, J. Chromatogr. A, 1510, 82–88.
15. Omar H. Ismail, Gioacchino L. Losacco, Giulia Mazzoccanti, Alessia Ciogli, Claudio VillaMartina Catani, Luisa Pasti, Scott Anderson, Alberto Cavazzini, and Francesco Gasparrini, “Unmatched Kinetic Performance in Enantioselective Supercritical Fluid Chromatography by Combining Latest Generation Whelk-O1 Chiral Stationary Phases with a Low-Dispersion in-House Modified Equipment”, 2018, Anal. Chem., 90, 10828-10836.
16.] Ruben De Pauw, Konstantin Choikhet, Gert Desmet, Ken Broeckhoven, “Occurrence of turbulent flow conditions in supercritical fluidchromatography”, 2014 J. Chromatogr. A 1361 277–285.
17.] Terry A. Berger, “Characterizing pressure issues due to turbulent flow in tubing, in ultra-fast chiral supercritical fluid chromatography at up to 580 bar”. 2016, J. Chromatogr. A, 1475, 86-94.
18.] Terry A. Berger. “High Speed, High Efficiency Achiral SFC on a 3x20 mm Column Packed with 1.8 µm Particles Facilitated by a Low Dispersion Chromatograph”, 2019, Chromatographia, 82, 537-542; DOI 10.1007/s10337-018-3655-5.
19.] Terry A. Berger, “Reduced Plate Height of 1.65 on a 20x3 mm column packed with 1.8 µmParticles in Supercritical Fluid Chromatography” Chromatographia, accepted April 2019.
20.] A.C. Sanchez, J.A. Anspach, T. Farkas, “Performance optimizing injection sequence for minimizing injection band broadening contributions in high efficiency liquid chromatographic separations”, J. Chromatogr. A 1228 (2012)338-348.
21.] James C, Sternberg, “Extracolumn Contributions to Chromatographic Band Broadening”, in Advances in Chromatography, Vol.2, J.C. Giddings, R.A. Keller, eds., Marcel Dekker, New York, 1966, Chapter 6.
22.] Martin Enmark, Dennis Åsberg, Andrew Shalliker, Jörgen Samuelsson, Torgny Fornsted “A closer study of peak distortions in supercritical fluid chromatography as generated by the injection” J. Cromatogr. A, 1400, 2015, 131-139.
23.] Vincent Desfontaine, Abhijit Tarafder,Jason Hill, Jacob Fairchild, Alexandre Grand-Guillaume Perrenoud,, Jean-Luc Veuthey, Davy Guillarme, “A systematic investigation of sample diluents in modern supercritical fluid chromatography” J. Chromatogr. A, 1511 (2017) 122-131