Supercritical fluid (SFC), green chromatography
Published over 7 years ago. See the latest and most current information on Supercritical fluid (SFC), green chromatography.
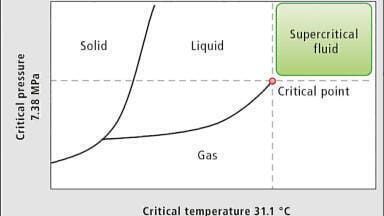
The history of supercritical fluid chromatography (SFC) has been fraught with controversies that largely held back the development of the technique for decades. Perhaps the greatest problem was the lack of any physical-chemical data, such as the density, viscosity, or compressibility of CO2-modifier mixtures, or solute diffusion coefficients under realistic mobile phase conditions. This lack of data resulted in numerous erroneous assumptions resulting in false starts and misdirection’s. The development of adequate hardware has also been an important issue, culminating in the last few years with true 3rd generation instruments. The present state of affairs is also briefly outlined, and some predictions about the future are made.
The Pioneers
Since the late 1800’s it has been known that some heavy, non-volatile organic compounds were surprisingly soluble in some inorganic gases above their critical point (‘super’-critical). These gases include SO2, CO2, ammonia, etc. It was erroneously thought that the fluids had to be above the critical point to act as a solvent. At an early gas chromatography (GC) conference in 1957, Jim Lovelock suggested using such inorganic gases, above their critical points as a chromatographic mobile phase, in order to separate much more polar compounds compared to GC. Acting as a solvent is an important characteristic that differentiates the compressible mobile phases used in supercritical fluid chromatography (SFC) from gas chromatography (GC), where the mobile phase is considered to be an inert carrier. Lovelock suggested the name ‘critical state chromatography’.
Ernst Klesper was the first to actually use a fluid above its critical point to separate thermally labile metal porphyrins. The first publication we can call SFC appeared in 1962, as a three page ‘Communications to the Editor’ in the Journal of Organic Chemistry [1], with no figures. He used chlorofluorocarbons at 800-2300 psi and 115°C which were pre-heated in a copper coil to generate high pressure (no pump), before passing through the 30 inch long column with a 60-80 mesh (180-250 µm) diatomaceous earth GC packing(!). There was no detector. After the column, the fluid was chilled to a liquid. Fractions were collected and analysed off-line. The elution strength was found to be proportional to pressure. Flow rate was not ‘controlled’, but was basically adjusted within a range, by replacing fixed restrictors, or using a metering valve.
From this beginning, there was, and still is, significant difficulty in coming up with an appropriate name that captures the essence of the technique in a short, simple phrase. Klesper called the technique ‘high pressure GC above critical temperatures’. Clearly this was a poor name since the mobile phase acted as a solvent and wasn’t inert. Thus, it isn’t GC.
Giddings, the most influential chromatographic theorist in the 1960’s used many names, including: turbulent flow chromatography [2], ultra high pressure gas chromatography (to 2000 atmospheres) [3], and dense gas chromatography [4]. None of these names captured the solvating nature of the mobile phase. Sie and Rijnders [5] first used the name supercritical fluid chromatography, but this wasn’t much better since this name implies the fluid must ALWAYS be ‘super’ (meaning above) the critical point to display the desirable characteristics. Caude [6] was apparently the first to use the term “subcritical” to denote the fact that some modified CO2 based fluids were of high density, while still highly compressible, but acted as a solvent even below their critical temperature. The idea that the fluids somehow drastically changed solvent power when passing from sub- to supercritical goes all the way back to 1874, and some still think this is true.
Early Controversies Retarded the Growth of SFC
In the late 1960’s Giddings estimated Hildebrand solubility parameters [7] which suggested that dense CO2 could be as polar as isopropyl alcohol (IPA). It was thought that programming pressure (or density) could adjust the elution strength of CO2 from that of a hydrocarbon to that of an alcohol! This was before the invention of high performance liquid chromatography (HPLC), and there was much that was still poorly understood. If this were true, SFC would today be as big, or bigger, than HPLC. Unfortunately, it isn’t true. Later, many used Giddings elutropic series to suggest adding a polar modifier, like methanol, to CO2, did not significantly increase solvent strength, but only covered active sites. Some even suggested the modifier increased the density of the mobile phase which we now know is only true over a narrow range of low modifier concentrations.
Another controversy began in the late 1960’s when Sie and Rijnders [5], Milos Novotny [8], Gouw and Jentoft [9], and later, L.B. (Buck) Rogers [10] all suggested that the efficiency of their very crude packed columns (up to 120µm packings) seemed to degrade with higher pressure drops. Gouw and Jentoft [9] suggested that decreasing density along the axis acted like a decreasing temperature gradient in GC, causing a loss in efficiency.
By the mid to late 1980’s, it was widely thought that pressure drops of as little as 20 bar seriously degraded efficiency. There were actually several competing theories [11,12] trying to explain why this should occur. It was claimed that SFC packed columns could never produce more than ≈20,000 plates [13], and virtually ruled out any use of particles smaller than 5µm. These theories were unrelated to, but contemporary with, the commercialisation of capillary SFC (see next section). This problem stemmed directly from inadequate home-made equipment, related to back pressure control and the use of fixed restrictors instead of a back pressure regulator. In many cases the column outlet pressure was not even monitored, allowing operation in inappropriate regions.
One additional controversy further retarded the use of packed columns through the 1980’s. Polar solutes often did not elute, or eluted with poor peak shapes even at high modifier concentrations. This was blamed on “active sites” on the silica based stationary phases. Widespread consensus was to make the phases more non-polar and increase end-capping to cover such sites. However, such columns resulted in minimal retention but still poor peak shapes. Even polymer-based columns, without silanols, produced similar poor peak shapes. The problem was largely solved when it was shown [14-19] that the introduction of polar additives, such as a strong acid or base, in the modifier, dramatically improved peak shapes for polar solutes. This is largely due to insuring the solutes have neutral charge, plus the additive competing for active sites. Many papers about the use of additives in SFC have followed, and dramatically expanded the application areas amenable to SFC. Today SFC brackets all the area amenable to both normal phase and reversed phase HPLC and further includes parts of ion chromatography, HILIC etc. Much more recently, new stationary phases have been developed based on ethylpyridine and imidazole chemistries (Princeton Chromatography and ES Industries respectively), specifically to allow improved peak shapes without an additive, to improve interfacing with mass spectrometric detectors.
Capillary or Open Tubular SFC
Due largely to the perceived limitations discussed in the previous sections, a major detour occurred starting around 1981 when Milton Lee and Milos Novotny first described capillary SFC [20], made possible by the recent, nearly concurrent inventions of fused silica capillary columns and bonded stationary phases. Their primary assumption was that significant pressure drops caused catastrophic efficiently losses, based on the results (including Novotny’s) from the 1970’s. By simply using a syringe pump to change the pressure of pure CO2, the solvent strength of the mobile phase was thought to be programmable from a hydrocarbon to an alcohol while also using the universal flame ionisation detector (FID). Such an approach seemed to solve all the problems associated with packed columns but was based on the unfortunate mis-step by Giddings, and the mis-interpretation of the effect of pressure drops on efficiency. The simplicity of such an approach was, for a time, overwhelming, and almost completely replaced packed column usage. There was very active research in the area by Karin Markides, Keith Bartle, Steve Hawthorn, and many others. At one point there were six or seven companies manufacturing and selling capillary SFC’s, and only one (Jasco) selling packed column SFC’s.
The tide had turned to such an extent away from packed columns and toward capillaries that David (Dai) Games, a prominent HPLC-MS and SFC-MS guru had a packed column talk rejected by Pittcon in the late 1980’s. He was reduced to passing out copies of his chromatograms at the door of the SFC meeting. Never the less some practitioners, such as Rodger Smith, and a large group at then Ciba-Geigy continued to publish packed column results.
The Return to
Packed Columns
When density measurements for methanol/CO2 mixtures were finally made in 1990 [21,22], it was shown that, at constant density, modifiers significantly increased the solvent strength of the mobile phase. Later, solvatochromic dye studies [23] unequivocally showed that CO2 was never much more polar than hexane, and modifiers dramatically increased solvent strength. Never the less, decades of belief are slow to be changed.
The idea that large pressure drops cause significant losses in efficiency and limited total possible efficiency was disproved in 1990’s when [24] many columns connected in series, produced 220,000 plates with a pressure drop of < 300 bar.
By the early 1990’s, the concentration of polar modifiers was shown to be the primary retention control variable while pressure/density became a secondary control variable. The use of additives allowed the elution of polar solutes, such as primary aliphatic amines with high efficiency. Thus, modern SFC was born, while capillary SFC almost completely died out.
Columns
All the work on packed columns up to 1980 used huge, irregular shaped particles, such as 120µm with a wide size distribution, suitable only for antique GC applications. They were often used in relatively short columns such as 30 inches. In SFC, the diffusion coefficients are several orders lower than in GC, meaning the chromatography with these columns was very slow with poor efficiency. The results obtained were extremely confusing, particularly with respect to pressure drops along the column. In this author’s opinion, it would have been far better if such results had never been collected since they distorted and continue to distort understanding.
This situation changed in 1980, when Dennis Gere, at Hewlett Packard, was the first to use ‘modern’ spherical, totally porous 3µm packings, which generated high back pressures but very high speeds and very high efficiency. Gere stated that as long as the pressure drops occurred away from the near-critical region, there was minimal to no loss of efficiency or speed. With this work it became clear that solute diffusion coefficients were 3 to 5 times faster in CO2 based fluids, and pressure drops were simultaneously 1/3rd to 1/5th, compared to HPLC. Since then, the packings used have been the same as in hplc.
It is now fairly common to use sub-2µm totally porous and superficially porous particles with various chemistries applied. The most common phases are normal phase, such as bare silica, but there are also many bonded phases available. For relatively non-polar solutes such as glycerides reversed phase columns such as C18 are applicable. This is sometimes called reversed phase SFC. However, the modifier remains more polar than the CO2 and retention decreases with increased modifier concentration, which is normal phase.
Instrumentation
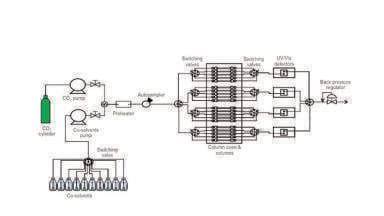
Until the early 1980’s all instrumentation was home-made, using components from various other fields. Many of the misconceptions of the 1960 through the 1980’s can be directly attributable to the poor instrumental controls then prevalent, particularly the poor control of outlet pressures. In many ways, reproducible SFC started with the introduction of a hardware kit that converted a Model 1084 HPLC into an SFC, introduced by Hewlett Packard (now Agilent) in 1982. This instrument was crude by today’s standards. Its main contribution was a heated mechanical back pressure regulator (BPR) controlling the column outlet pressure, with large mechanical gauges unequivocally showing both the inlet and outlet pressures. Retention control rested entirely on the mobile phase composition. This instrument was withdrawn around 1985, when the Model 1090 HPLC was introduced which was incompatible with SFC. Jasco introduced a SFC-SFE combined instrument in 1985. This instrument included the first electronic BPR, but still did not perform dynamic compressibility compensation, meaning the actual volumetric flow and the composition changed with increasing pressure.
By the early 1990’s capillary SFC had been grossly oversold, particularly for the separation of more polar drug-like solutes. This ‘poisoned the well’ for about ten years where many regarded SFC as ‘science fiction chromatography’. Fortunately the clear superiority of packed column SFC over HPLC for chiral analysis was obvious and, just sustained the small start-up manufacturers through this period.
In 1992 several second generation SFC’s capable of packed column operation were introduced by Hewlett Packard (HP), and Gilson, although only HP used dynamic compressibility compensation. Proper compressibility compensation means that both the flow rate and composition (v/v%) were for the first time, accurate, making transferring methods from machine to machine easier. This also marked the first Peltier cooled pump head, a return to pressure, density and temperature programming as well as composition programming. Electronic back pressure regulators became the norm on virtually all subsequent SFC’s. Diode array detectors significantly extended the utility of UV detectors. Many GC detectors, such as the flame ionisation (FID), electron capture (ECD), and nitrogen-phosphorus (NPD), etc. were also commercially available. GC like ovens allowed for much higher column temperatures, necessary for the separation of many oligomeric samples such as surfactant, silicone oils, hydrocarbons, etc. Today, air bath ovens are clearly a bad idea since they promote radial temperature gradients [25], resulting in efficiency losses.
Packed column SFC started to gain traction in the early 1990’s with the separation of enantiomers. The first use of a chiral stationary phase was by Caude in 1985 [6]. The first SFC use of a chiral additive in the mobile phase was by Erni [26] in 1988. Today, far and away, the ‘killer app’ for SFC has been in chiral chromatography at both the analytical and semi-preparative scale. SFC is easily 3-5 times faster than HPLC, has much lower pressure drops, is less expensive to operate, generates much less toxic and flammable waste (more ‘green’), and is much easier to dry down fractions. Consequently, in many major pharmaceutical companies SFC has become the technique of choice for these applications, whereas HPLC has been relegated to a few difficult cases [27], or has been eliminated from use altogether [28].
The Present
The present probably started in 2009 with the introduction of the Aurora SFC conversion module that converted an Agilent Model 1100 or Model 1200 HPLC into a world class SFC. Waters introduced its SFC as Ultra Performance Convergence Chromatography (UPC2) in 2012. Agilent bought Aurora in 2012 and better integrated the module into the 1200 series of HPLC’s. Shimadzu introduced a combined SFC/SFE a few years later. Jasco has upgraded its SFE/SFE several times over the last 20 years. These instruments are true third generation SFC’s, with superior performance, although none have the versatility of some earlier instruments. The main improvement was in better UV noise. More importantly, it means that for the first time four of the largest instrument companies are committed to selling and supporting SFC, worldwide. This is a major change from the past. From 1995 until ~2007 the majority of SFC’s sold were produced by small start-up companies with limited resources. It worked, in large part, due to the stunning superiority of SFC over HPLC in chiral analysis and purification.
For clarification, the current generation of commercial SFC’s are NOT true ultra high performance SFC’s. ‘Ultra’ performance is defined here as producing >90% of theoretical efficiency with a k of ≥2 using sub-2 µm particles. The current commercial plumbing is similar to standard HPLC’s from 10+ years ago, in that the extra-column variance is on the order of ≈85-90µL2 [29]. Modern UHPLC’s have variances of a few µL2. Fortunately, it is fairly easy to modify some of the equipment to allow use of sub-2µm particles in 3mm ID columns [30], although most users seem reluctant to implement such changes. It is hoped that any future instrument designs include much shorter lengths of connector tubing with smaller volume, but hopefully long pathlength, low volume, UV detector cells, such as used in some UHPLCs.
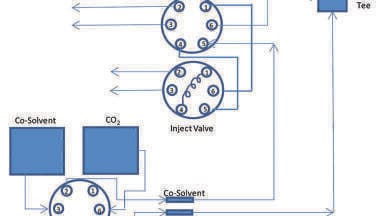
Agilent uses the conversion module mounted upstream of a nearly standard HPLC pump to pre-compress the CO2 to just below column head pressure. The compressibility compensation for the HPLC pump delivering CO2 is then set to zero. Thus, this pump merely meters the CO2 flow, virtually eliminating any flow/pressure fluctuations from the CO2 pump, a major source of UV noise in previous SFC’s. This, in combination with an ultra low noise back pressure regulator (BPR), and careful control of mobile phase temperature entering the detector cell, resulted in as much as a 50 fold decrease in UV detector noise compared to earlier instruments.
Waters uses a slightly different approach to deal with compressibility compensation related noise. Each piston of a two piston pump (delivering a single fluid) is driven by a separate motor, much like the old Rainin SD-1 pumps. While the first piston is delivering flow, the other refills, then pre-compresses the fluid to just below column head pressure, and then waits. When the first piston reaches the end of its delivery stroke, it slows down and passes off delivery to the already refilled and pre-compressed pump head. Since the compression stroke is completely independent of the delivery stroke there is minimal flow/pressure/composition noise, similar to the Agilent approach. Both use two motors to pre-compress the CO2, just differently.
The dramatic improvement in UV noise means that, for the first time, SFC was appropriate for trace analysis where it can easily quantify peaks representing 0.1% (area/area) of a major component, with a signal to noise ratio >10. This opens up the possibility of using SFC in quality assurance (QA), and quality control (QC). In the past, the poor sensitivity relegated SFC to major/minor component analysis (not trace), mostly in drug discovery. The possibility of also performing routine and trace analysis should dramatically expand the use of SFC.
Although not mentioned previously, SFC has been widely used with mass spectrometric detectors (MS) in all its forms. Virtually every MS instrument and interface has been used with SFC over the years, including the most common ionisation sources such as atmospheric pressure chemical ionisation (APCI), electrospray ionisation (ESI) and many others. SFC has been used with simple quadrupole instruments as well as MS/MS, MS/MS/MS and QTOF. Today the interfaces have become trivial, sometimes no more than a specifically sized stainless steel tube.
The Future
It is likely that all SFC’s will eventually use pumping systems that separate the compression stroke from the delivery stroke, since this can dramatically decrease UV detector noise, and reduce or largely eliminate gradient delays. Such pumping systems are common in UHPLC and will probably be the norm in SFC soon.
Due to the low viscosity of SFC mobile phases, sub-2 µm particles only generate < 250 bar column pressure drops, even at 40% methanol and 5ml/min (3mm ID, 100mm long column). However, a recently recognised issue in SFC arises from the low viscosity and high flow rates required in SFC, compared to HPLC. In smaller ID tubing (≈125µm), flows above ≈3ml/min produce turbulent flow [31,32]. With turbulent flow, the P increases more than the inverse square of tubing ID. The P in the connector tubing is often much larger than the column P. Some of this pressure drop occurs before and some after the column. Unlike in HPLC, SFC retention is partially dictated by mobile phase density/pressure. With a large pressure drop in front of and behind the column the pressure/density in the column becomes progressively more difficult to determine. In HPLC, the pressure drop in the connector tubing is irrelevant to retention so the solution is to use much smaller ID tubes such as 75 or 100µm and simply increase the pump pressure.
Using such tubes in SFC will result in extreme pressure drops outside the column even at lower flow rates, and will likely require pump pressures well above 600 bar, while further obscuring the actual pressure/density in the column.
Since the use of very small ID connector tubing, used in UHPLC, isn’t viable in SFC, the obvious alternate solution is to abandon the architecture of discrete modules performing each discrete function, such as pumping, injection, column temperature control, and detection each module connected to the next by long connection tubing. With a more integrated design, the connector tubing lengths could be shortened dramatically and extraneous pressure drops significantly reduced. However, funding a hardware development that deviates from UHPLC, may be problematic.
Recently a major change in HPLC has involved the use of very short (0.5-3mm) well packed columns with sub-2µm particles, particularly in chiral analysis. There are now chiral HPLC separations performed in under 1 sec. Although extremely limited in scope, such extremely fast UHPLC makes irrelevant the traditional advantage that SFC is 3-5 times faster than HPLC. SFC can be similarly fast, or probably faster, but is such differentiation relevant? It appears that in some cases HPLC and SFC will become techniques for on-line process monitoring. This has significant implications for the need for instrument and software change. It also suggests a path toward fast 2-D, 3-D HPLC-SFC, and MS combinations.
Since semi-prep SFC is far superior to HPLC in terms of speed, pressure drops, greenness, etc. analytical scale SFC will continue to be a major asset for chiral separations in major pharma. Amazingly, most academics and smaller organisations continue to ignore chiral SFC, possibly due to the higher entry cost to buy the equipment, or the perceived lack of versatility. It is hoped that the greater marketing capabilities of the major instrument companies now involved can get the message across that much better, faster results are possible. One of the biggest obstacles for SFC growth in the past has been the very limited teaching of the technique in universities. This remains an obstacle, particularly in the United States. In Europe there is a surprisingly large number of universities using SFC.
SFC is now 10-50 times more sensitive than previous generation instruments and capable of validation for the quantification of 0.1% area components in complex mixtures with a S/N >10, with the same sensitivity as HPLC. The mobile phase used in SFC is generally cheaper, and ‘greener’ than HPLC.
SFC is orthogonal to HPLC. It is usually necessary to have two methods for trace analysis to prove that a trace component was NOT eluting under a major component in the first method. In HPLC, normal phase is so slow and difficult that two reversed phase methods, with minimal differences, have often been used. SFC is normal phase but actually faster than reversed phase HPLC. Thus, there is no compromise when using a normal phase (SFC) technique, based on polar-polar interactions, and a reversed phase technique based on hydrophobic interactions. In fact, commercial hybrid systems are available that can switch back and forth between SFC and reversed phase HPLC in only a few minutes.
SFC is likely to be used to a much greater extent than in the past in food, fuels, and natural products in both research and in routine analysis. Fields other than pharma, including pesticides, are likely to perform many more chiral analyses. The ‘green’ aspects of SFC are likely to become more widely appreciated.
References
1. E. Klesper, A.H. Corwin, D.A.Turner, (1962) J. Org. Chem., 27 700-706
2. J.C. Giddings, W.A. Manwaring, M.N. Myers, (1966) Science 154,146-148.
3. M.N. Myers, J.C. Giddings, (1966) Sep.Sci., 1,761-776.
4. J.C. Giddings, M.N. Myers, J.W. King, (1969) J. Chromatogr. Sci. 7,276-283.
5. S.T. Sie, and G.W.A. Rijnders, (1967) Sep Sci., 2, 729-753.
6. P.A. Mourier, E. Eliot M.H. Caude, R.H. Rosset, (1985) Anal. Chem., 57,2819-2823.
7. J.C. Giddings, M.N. Myers, L.M. McLaren, R.A. Keller, (1968) Science, 162 67-73
8. M. Novotny, W. Bertsch, A. Zlatkis, (1971) J. Chromatogr. 61, 17-28.
9. T.H. Gouw, R.E. Jentoft, (1972) J. Chromatogr. 68, 303-323.
10. J.A. Graham, L.B. Rogers, (1980) J. Chromatogr Sci., 18, 75
11. P.J. Schoenmakers C.C.J.G. Verhoeven , (1986) J. Chromatogr., 352, 315-328.
12. P.A. Mourier, M.H. Caude, R.H. Rosset, (1987) Chromatographia, 23, 21-25.
13. P.J. Schoenmakers, ‘Open Column or Packed Columns for Supercritical Fluid Chromatography’, in Supercritical Fluid chromatography RM Smith ed. RSC Chromatography Monographs. The Royal Society of Chemistry, London, UK 1988, Chapter 4.
14. M. Ashraf-Korassani, M.G. Fessahaie, L.T. Taylor, T.A. Berger, and J.F. Deye, (1988) J. High Resolut. Chromatogr., 11, 352
15. T.A. Berger, J.F. Deye, M. Ashraf-Korassani and L.T. Taylor, (1989) J. Chromatogr. Sci., 27, 105-110
16. T.A. Berger, J.F. Deye, (1991), J. Chromatogr. Sci., 29,26-30.
17. T.A. Berger and J.F. Deye, (1991) J. Chromatogr. Sci., 29,141-146.
18. T.A. Berger and J.F. Deye, (1991) J. Chromatogr. Sci., 29,310-317.
19. T.A. Berger, and J.F. Deye, (1991) J.Chromatogr., 547,377-392.
20. M. Novotny , S.R. Springston , P.A. Peaden , J.C. Fjeldsted , M.L. Lee, (1981) Anal. Chem., 53,407A–414A.
21. T.A. Berger, (1991) J. High Resolut. Chromatogr., 14, 312-316.
22. T.A. Berger , J.F. Deye, (1990) Anal. Chem., 62, 1181–1185
23. J.F. Deye, T.A. Berger, A.G. Anderson, (1990) Anal. Chem., 62 615-622
24. T.A. Berger, W.H. Wilson, (1993) Anal. Chem., 65, 1451–1455.
25. Sam O. Colgate, Terry A. Berger, J. Chromatogr. A, 1385, 2015, 94-102
26. W. Steuer, M. Schindler, G. Schill, F. Erni, (1988) J. Chromatogr. A, 447,287-296.
27. C. White, J. Chromatogr. A, 1074 (2005) 163-173.
28. M. Maftouh, C. Granier-Loyaux, E. Chavana, J. Marini, A. Pradines, Y. Vander Heyden, C. Picard, (2005) J. Chromatogr. A, 1088, 67-81.
29. Alexandre Grand-Guillaume Perrenouda, Chris Hammanb, Meenakshi Goelc,Jean-Luc Veutheya, Davy Guillarmea, Szabolcs Feketea,, Journal of Chromatography A, 1314 (2013) 288– 297
30. Terry A. Berger. J. Chromatogr. A, 1444 (2016) 129-144.
31. Terry A. Berger, J. Chromatogr. A, 1475 (2016) 86–94.
32. Ruben De Pauw, Konstantin Choikhet, Gert Desmet, Ken Brockhoven
J. Chromatogr. A, 1361, 2014, 277- 285.