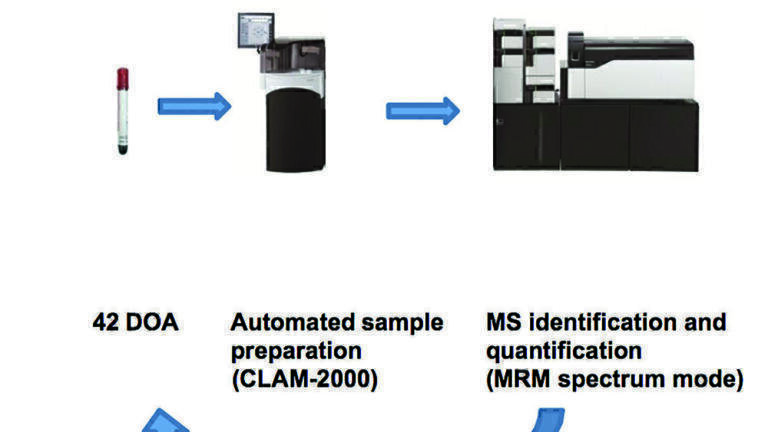
Sample preparation
Published over 8 years ago. See the latest and most current information on Sample preparation.
For the analysis of drugs and pharmaceutical compounds in biological matrices, LC-MS/MS analysis is typically preceded by sample preparation which often requires manual steps. In this study, we report a fully automated extraction process directly coupled to an LC- MS/MS system for the determination of amphetamines, cocaine and opiates.
42 target compounds and 20 deuterated internal standards were included in the method. The extraction was carried out by a programable liquid handler (CLAM-2000, Shimadzu) coupled directly to an LC-MS/MS system (Nexera X2 & LCMS-8060, Shimadzu). The acquisition was performed in positive ionisation mode with up to 15 MRM transitions per compound, each with optimised collision energy (Multiple Reaction Monitoring or MRM Spectrum mode) to enable qualitative library searching in addition to quantitation.
This approach is successfully designed to support parallel sample preparation and analysis therefore significantly increasing sample throughput and reducing cycle times.
Introduction
Opiates, amphetamines (including analogues) and cocaine are widely used drugs of abuse (DOA) and many laboratories have developed LC-MS/MS procedures to identify and quantify these compounds [1-5]. Such measurements are needed in multiple contexts within clinical and forensic toxicology (suspicion of overdose, monitoring of addicts, driving under the influence of drugs, doping control and pain relief).
To minimise the possibility of false positive or negative results reporting without compromising accuracy, precision and limits of detection, methods were developed to combine the sensitivity of MRM detection with the identification power of MRM spectrum. Contrary to an entire mass spectrum, MRM represents the operation of the mass spectrometer in which a precursor ion is selected and then the abundances of multiple product ions are recorded. In the present study, the method has the capability of following up to 15 MRM transitions per compound and enabling precise, accurate quantitation and library searchable compound identification. Each transition has optimised collision energies for each ion. Ion intensities from each transition are used to construct an MRM Spectrum that can be used to search against registered library spectra.
To develop an automated generic sample preparation method in clinical toxicology analysis, an automated sample preparation system was coupled to LC-MS/MS system. Once the primary tube is loaded onto the intervention was required. The sample is automatically delivered to a tube containing a filter, to which reagents are added, mixed and then filtered. The extract is finally injected into the LC-MS-MS system.
The procedure was fully validated which included: repeatability, reproducibility, matrix effects, extraction yields, inter- matrix agreement, dilution tests and robustness.
To test its viability, the method was applied to patients’ blood or plasma samples and compared against a validated LC-MS/MS method using 2 MRM’s for each target compound [5].
Experimental
The sample is automatically delivered to a tube containing a filter, to which reagents are added, mixed and then filtered.
Precisely, 100 µL of acetonitrile were added to a PTFE filter vial (0.45 µm pore size) previously conditioned with 20 µL methanol. Then, 50 µL of plasma (or whole blood) and 10 µL of isotopically labelled internal standards (0.2 mg/L in acetonitrile) were added. The mixture was mixed for 120 seconds (1900 rpm) then filtered by application of vacuum pressure (-60 to -65 kPa) for 120 seconds into a collection vial. Finally, 3 µL of the extract was injected into the LC-MS-MS system.
All compounds were measured by scheduled MRM, with up to 15 transitions per compound throughout the entire scheduled window using 1 msec pause time and 3 to 10 msec dwell time. All transitions were collision energy optimised using flow injection analysis. Chromatographic peak apex intensity was used to extract ion intensities for construction of an MRM Spectrum.
Validation and Robustness Study
The laboratory of Pharmacology-toxicology of the Limoges University Hospital works towards accreditation by the International Standards Organization (ISO) 15189 standard (accreditation number: 8-2607). These requirements were applied to the present method.
The intra-assay precision and accuracy (n=6) and the inter-assay and accuracy (n=6) were assessed at lower limit of quantitation (LLOQ; 1 or 2,5 or 5, depending on the compound), 50 and upper limit of quantitation (ULOQ 500 ng/mL) after complete extraction procedure and analysis of six different spiked plasma samples (compound-free human plasma) for each level. To assess the inter-assay precision and accuracy, a set of calibrating samples was analysed each day for 6 days. The lower limit of quantitation (LLOQ) was defined as the lowest concentration of compound that could be measured with both an intra-assay and inter-assay precision (CV%) and an accuracy (bias) less than 15%.
Calibration standards were prepared by adding automatically appropriate working standard solutions to 50 µL of plasma prior to extraction in order to obtain concentrations ranging from 1 to 500 ng/mL for all compounds (6 levels, 1 or 2,5 or 5 depending on the compound, 10, 50, 100, 200 and 500 ng/mL). Calibration graphs of the compounds of interest-to-internal standard peak-area ratios of the quantification transition versus expected compounds of interest concentration were constructed using a quadratic with 1/x or 1/x² weighting regression analysis.
Recoveries were determined at two concentration levels (LLOQ and 500 ng/mL) by comparing the analyte / internal standard peak area ratios obtained after extraction of spiked samples (n=6) with those of DOA-free plasma extracts further spiked with the DOA (n=3).
The effect of dilution was investigated using samples manually spiked at 150% of ULOQ then re-analysed after 2, 4 and 10-fold dilutions.
The absence of carry over was checked.
Matrix agreement was tested for whole blood: the intra-assay precision and accuracy were assessed by preparing six replicates at LLOQ and at 500 ng/mL in whole blood from 6 different individuals and calculated with a calibration standard prepared in plasma. Precision CV and bias were set less than 15% to successfully validate.
Matrix effects were evaluated in whole blood and plasma, for the 42 molecules. Six different whole blood and plasma samples were tested. In each case, potential ion suppression or enhancement was explored by comparing the signal observed for the molecule of interest (whole blood or plasma spiked at 50 ng/mL) to that obtained with Milli Q water/acetonitrile (1/2).
A robustness study was performed to evaluate the acceptable quantitative accuracy that could be provided by a calibration curve. Freshly prepared control standards (5 and 50 ng/mL) were quantified with freshly prepared calibration standards over a 4 week period. Control sample data were first processed using calibration standards prepared on the same day as the control samples and then re-processed using calibration standard data which are up to 4 weeks old.
Results
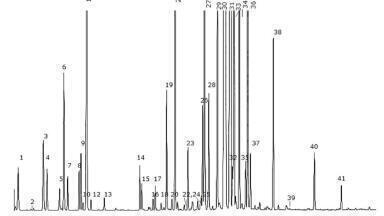
Automated sample preparation was performed in 8 minutes followed by chromatographic separation of the DOA in about 9 min (with an additional 9 min for column wash and equilibration) with retention time from 0.97 min for ecgonine methylester to 7.9 minutes for methadone. About 26 minutes were needed to obtain the first result and then, extraction and separation were performed in parallel with the system producing a result every 18 minutes. Table 3 summarises the results of the optimisation of MRM transitions acquisition. Up to 15 MRM transitions were obtained for a targeted compound.
The results of the validation study are summarised in Table 4. Acceptance criteria were obtained for all explored parameters. Concerning the intra-assay and the inter-assay (n=6) precision and accuracy, the CV values were less than 15% (except for benzoylecgonine, cocaethylene, EDDP and naltrexone for which values less than 20% were reported at the LLOQ). Using quadratic models with a 1/x or 1/x² weighting regression, the correlation coefficients of the calibration curves (LLOQ to 500 ng/mL) were higher than 0.99 for all the compounds. Depending on the molecule, the LLOD and the LLOQ were set at 1, 2.5 or 5 ng/mL. No matrix effects (n=6) were reported in our conditions. Dilution tests (n=3) also reported good results.
The quantification of the control standards (5 and 50 ng/mL) with calibration curve acquired up to one month before the injection of the controls produced accuracy variation between 70 and 130%. The maximum CV value was 13.0% for the control at 5 ng/mL and 14.9% at 50 ng/ml. Correct accuracy was also obtained for the quantification of the control standard with calibration curve acquired up to one month after the injection of the controls. The maximum CV was 13.4% for the control at 5 ng/ml and 14.2% at 50 ng/mL.
Figure 2 illustrates the approach for 2 isobaric compounds.
Application of the Whole Procedure to Patient Sample(s)
The whole automated sample preparation and LC-MS/MS analysis was tested by comparing quantitative results from 43 patients samples (plasma or whole blood) prepared by the automated technique with those from a pre-existing validated method using an LCMS-8050 system, using QuEChERS salts extraction method, routinely used in the lab [5]. The automated sample preparation method was measured by MRM Spectrum mode whereas the LCMS-8050 system measured samples using a conventional MRM method. Patient blood or plasma samples were obtained from a diverse range of backgrounds commonly encountered in the laboratory including routine drug testing, DUID or emergency overdose.
Figure 3 shows a global agreement in terms of quantitation of these compounds.
Conclusion
We report a fully automated LC-MS/MS analysis method for the detection and determination of DOA in blood with the inclusion of library identification using MRM Spectrum mode.
The implementation of automation for all or part of the analysis process eliminates human errors made by manual preparation and saves time in the laboratory enabling technicians to perform other manual tasks while the system performs the analysis automatically.
We have developed a method where no human intervention was necessary when the primary tube was loaded on board the system. Sample preparation was synchronised with the LC-MS/MS system resulting in no time being lost whilst maintaining the ability to prepare the sample on- line and direct injection immediately after preparation.
A spectral acquisition method was used that allows a reconstruction of a spectrum containing all the specific transitions of a molecule. Unlike other previously published approaches where two or three collision energies were applied to all molecules in a method using product ion scanning, the collision energy for up to 15 transitions per molecule have been optimised. This approach makes it possible to obtain extremely specific and rich spectral information. Furthermore, no threshold triggering was applied, so all MRMs were measured during the entire scheduled acquisition period. Therefore, even at very low signal intensities an MRM Spectrum could be generated. By using very fast dwell and pause times the burden of measuring additional MRM transitions did not alter the sensitivity compared to the standard 2-3 transition approach and the 42 molecules were all validated to the requirements of ISO 15189 accreditation. Validation included: specificity, sensitivity and robustness of this method for the analysis of 42 DOA and we compared its performance with that of a method accredited in the laboratory in a panel of samples obtained from patients. Investigation in to the system stability and robustness by repeat calibration curve analysis demonstrated excellent reproducibility.
With inclusion of spiked deuterated standards in unknown samples for quality control purposes we estimated our results could be quantified with an uncertainty of less than 20% using a calibration curve dating up to one month. In the case of emergency patient sample analysis, quantifying a concentration from an unknown sample to this level of accuracy with such speed may mean that lifesaving treatment might be administered within a time frame which is normally not possible with conventional sample treatment and analysis.
References
1. Cailleux A, Le Bouil A, Auger B, Bonsergent G,Turcant A, Allain P. Determination of opiates and cocaine and its metabolites in biological fluids by high-performance liquid chromatography with electrospray tandem mass spectrometry. J Anal Toxicol. 1999;23: 620–4.
2. Moeller MR, Steinmeyer S, Kraemer T. Determination of drugs of abuse in blood. J Chromatogr B Biomed Sci Appl. 1998;713:91–109.
3. Saussereau E, Lacroix C, Gaulier JM, Goulle JP. On-line liquid chromatography/tandem mass spectrometry simultaneous determination of opiates, cocainics and amphetamines in dried blood spots. J Chromatogr B. 2012;885–886:17.
4. Anzillotti L, Odoardi S, Strano-Rossi S. Cleaning up blood samples using a modified “QuEChERS” procedure for the determination of drugs of abuse and benzodiazepines by UPLC–MSMS. Forensic Sci Int. 2014;243:99–106.
5. Dulaurent, S., El Balkhi, S., Poncelet, L., Gaulier, J.M., Marquet, P., Saint-Marcoux, F. (2016) QuEChERS sample preparation prior to LC-MS/MS determination of opiates, amphetamines, and cocaine metabolites in whole blood, Analytical and Bioanalytical Chemistry, 408(5), 1467–1474.
6. Sauvage, F.L., Gaulier, J.M., Lacha tre, G., Marquet, P (2008) Pitfalls and Prevention Strategies for Liquid Chromatography– Tandem Mass Spectrometry in the Selected Reaction–Monitoring Mode for Drug Analysis, Clinical Chemistry, 54:9, 1519–1527