Sample preparation
Published over 6 years ago. See the latest and most current information on Sample preparation.
A new HPLC method has been developed as an alternative to existing pharmacopeia methods for the assay determination of Salmeterol and Fluticasone propionate Inhalation Powder. The chromatographic separation utilises an isocratic elution in which mobile phase consisting of a buffer potassium dihydrogen phosphate (pH 3.0) and acetonitrile at 1.5 mL min−1 flow rate, 40ºC column temperature, 25ºC tray temperature and gradient wavelength UV detection between 210-239 nm. A stainless steel column (15cm x 4.6mm, 5µm) packed with octadecylsilyl silica gel (Hypersil BDS) was employed.
Being validated in accordance with ICH guidelines, this method provides a safer and easier solution for assay determination of Salmeterol and Fluticasone propionate compared to pharmacopeia methods. The major benefits of the new method are; using a wavelength gradient for the quantitation of Salmeterol and Fluticasone propionate which give max. UV absorptions at different wavelengths, using a simple buffer solution that is prepared simply and which is non-toxic to the analyst.
1. Introduction
Being developed as a potent β2-adrenoceptor agonist and having the long acting bronchodilator profile, Salmeterol Xinofotate (SX) is used to open the airways in the lungs to make breathing easier in the treatment of asthma and chronic obstructive pulmonary disease known as COPD [1].
Largely used as an inhaled corticosteroid (ICS), Fluticasone propionate (FP) is commonly used in combination with Salmeterol Xinofoate [2], forming an inhalation product consisting of a long-acting beta2-adrenoceptor agonist (LABA) plus a corticosteroid [3]. It is proven that the twice daily therapy of combining Salmeterol and Fluticasone Propionate is more effective than the monotherapy of inhaled corticosteroids alone particularly in terms of enhancing lung function and reducing asthma symptoms. Moreover, as mentioned by McKeage and Keam, the combination of Salmeterol and Fluticasone Propionate ensures a powerful, strongly tolerated choice in the maintenance and treatment of asthma [3].
Cyplos 50/500 mcg Powder for Inhalation (Arven, Turkey) was developed as a fixed dose combination of Salmeterol and Fluticasone Propionate. For the assay determination of Salmeterol and Fluticasone Propionate Inhalation Powder, there are several assay methods provided by several authorities.
The US pharmacopeia (USP) published a pre-dispensed monograph for Fluticasone Propionate and Salmeterol inhalation powder. According to this monograph the assay method shows the separation of FP and SX with an octadecylsilyl bonded silica gel 5cm x 4.6mm, 3.5µm column with an isocratic elution of 0.01 M sodium dodecyl sulfate, methanol and acetonitrile at a flow rate of 2.0 mL/min. The column temperature is 40ºC. Fluticasone Propionate UV is detected at 239 nm wavelength and Salmeterol detected using Fluorescence (FLR) detection with an excitation wavelength of 225 nm and a resulting emission of 305 nm [4].
The monograph for Fluticasone Propionate and Salmeterol Inhalation Powder published by British Pharmacopeia (BP) states that FP and SX are separated by a method using a flow rate of 1.5 mL/min, with a stainless steel column (20 cm x 4.6 mm) packed with 5 μm octadecylsilyl silica gel at 40ºC. The method proposes a detection wavelength of 239 nm and a fluorescence detection with an excitation wavelength of 225 nm and an emission wavelength of 305 nm. The proposed mobile phase contains acetonitrile, methanol and a solution containing 0.2M ammonium acetate and 0.5% w/v tetrabutylammonium hydrogen sulphate in water [5].
The assay method provided by USP 41 employs Sodium Dodecyl Sulphate, a surfactant in the mobile phase preparation that is known to be flammable, harmful if swallowed, causes skin irritation and serious eye damage plus many more hazards [6]. Moreover, in the USP method and the method provided by the BP, FLR detector is used, which while being quite sensitive to any contamination caused by working conditions. Any contamination detected by this detector is exhibited as an unknown peak in the chromatogram.
The new suggested method developed and proposed here utilises Potassium Dihydrogen Phosphate which is not classified as hazardous [7] instead of Sodium Dodecyl Sulphate. In addition, UV detection is used which is more common and shows fewer working condition contamination peaks compared to FLR detection. Furthermore, UV detection causes no selectivity difference in terms of the SX and FP peaks.
The purpose of this manuscript is to explain the assay method developed by Arven Pharmaceuticals for SX and FP fixed dose combination drug product, Cyplos 50/500 mcg Powder for Inhalation.
2. Materials and Methods
2.1. Reagents
SX, FP and their related impurities were purchased from a reputable API producer. Ortho-phosphoric Acid and Potasium Dihydrogen Phosphate Monohydrate were purchased from Merck Ltd. HPLC grade Acetonitrile was purchased from J.T.Baker. High purity deionised water was obtained from a Sartorius stedim, arium 611VF (Goettingen, Germany) purification system. Cyplos 50/500 mcg Salmeterol/Fluticasone Propionate Powder for Inhalation (Arven, Turkey) was used as the finished product. All impurities were European Pharmacopeia impurities as stated in Table 1.
2.2. Instrumentation
A Waters HPLC system consisting of inbuilt autosampler and quaternary gradient pump with an on-line degasser was used. The column compartment with temperature control and UV detector were engaged. Empower software was used to obtain chromatographic data.
2.3. Chromatographic Conditions
Equipment: HPLC System
Column: Thermo BDS
Hypersil C18
150 x 4.6 mm, 5 um
Flow Rate: 1.5 mL / min
Injection Volume: 40 µL
Column
Temperature: 40ºC
Tray Temperature: 25ºC
Run Time: 10 mins
Wavelength: UV, 210 nm
(Gradient Starting
Wavelength)
Wavelength gradient:
A Thermo Fisher Hypersil BDS C18 (15 cm x 4.6 mm) 5 µm column was used as stationary phase and maintained at 40ºC.
The Injection volume was 40 µL and the initial wavelength was 210 nm. In the analysis a wavelength gradient was applied representing the maximum wavelength response for SX and FP in Cyplos 50/500 mcg Salmeterol/Fluticasone Propionate Powder for Inhalation given above.
2.4. Preparation of Solutions
2.4.1. Mobile Phase
The mobile phase involved a fixed composition solvent A (pH 3.0 buffer) which was prepared by dissolving 1.0 g Potassium dihydrogen phosphate in a 1000 mL volumetric flask containing high purity deionised water and stirred at 1000 rpm for 20 minutes on a magnetic stirrer. The pH was adjusted to 3.00 ± 0.05 with 85% Ortho-phosphoric acid and stirred for a further 10 minutes.
Solvent A: Acetonitrile (520: 480 v / v) was prepared. Stirred for 15 minutes at 1000 rpm in a magnetic stirrer. The mixture was filtered through a 0.2 µm membrane filter and degassed for 2 minutes. The mobile phase was pumped through the column with a flow rate of 1.5 mL/min.
A Diluent mixture of acetonitrile : high purity deionised water (50:50, v/v) was also prepared for standard solution preparation.
2.4.2. Standard Solutions
Salmeterol Stock Standard Solution: ~14.53 mg Salmeterol Xinafoate standard (equivalent to 10.0 mg Salmeterol) was weighed and transferred into a 100 mL amber coloured volumetric flask and then diluted to volume with diluent stated above and dissolved by sonication in an ultrasonic bath for 15 minutes. The stock solution was then permitted to come to room temperature. (CSalmeterol = 0.1 mg/mL)
Fluticasone Propionate Stock Standard Solution: ~25.00 mg Fluticasone Propionate standard was weighed and transferred into a 100 mL amber coloured volumetric flask and then diluted to volume with aforementioned diluent. It was dissolved and dissolved by sonication in an ultrasonic bath for 15 minutes and allowed to come to room temperature. (C Fluticasone propionate = 0.25 mg/mL)
Standard Solution: 1.25 mL of the standard stock solution of Salmeterol and 5.0 mL of the standard stock solution of Fluticasone Propionate, transferred into a 50 mL amber coloured volumetric flask. The solution was made up to volume with dilution solution and mixed by shaking. The solution was filtered through 0.45 µm filter to a sample vial. (CSalmeterol = 0.0025 mg/mL, C Fluticasone propionate = 0.025 mg/mL)
2.4.3. Test solution from blister
Approximately 13.0 mg Cyplos 50/500 mcg Powder for Inhalation (containing 0.05 mg SX and 0.5 mg FP) was weighed in a 20 mL amber colour volumetric flask and made up to volume with dilution solution and dissolved by sonication in an ultrasonic bath for 15 minutes. The solution was then filtered through a 0.45 µm filter into a sample vial. (CSalmeterol = 0.0025 mg/ mL, C Fluticasone propionate = 0.025 mg/ mL)
2.5. System Suitability
The relative standard deviations for SX
and FP peak areas of six replicate injections of standard solution should not be more than 2.0%.
2.6. Optimisation of the
chromatographic conditions
Optimising the reverse phase HPLC parameters, several chromatographic conditions were tested in order to achieve a suitable peak resolution and peak shape for SX and FP.
2.6.1. Column Selection
Injections on to different columns types were conducted to achieve the best separation for the analyte peaks and other interfering blank and placebo peaks. The optimum peak shape, retention time, tailing factor, and column efficiency was achieved using a Hypersil BDS C18 column (15 cm x 4.6 mm, 5 µm).
2.6.2. Mobile Phase Composition
Different compositions of mobile phase were tested to obtain sufficient selectivity and retention time for the analyte peaks. With ammonium dihydrogen phosphate buffer, high sensitivity and selectivity were achieved when compared with other buffers. Based on peak shape, symmetry, retention time and peak tailing, pH 3.0 Potassium dihydrogen phosphate was selected as the buffer preparation to be used. Different gradient programs of pH 3.0 Potassium dihydrogen phosphate buffer and organic solvents were conducted and according to experiments with acetonitrile and methanol, higher retention time, higher column pressure and higher peak tailing were observed with methanol. Hence, acetonitrile was selected as the organic modifier. After many trials, based on the peak shape, peak symmetry, retention time and peak tailing a 1.5 mL/min flow rate was selected.
2.6.3. Detection Wavelength
SX, FP and their related substance peaks were scanned between 200 nm – 400 nm by photo-diode array detector. The maximum absorption of SX and FP was determined at 210 nm and 239 nm. Therefore, to obtain the maximum absorbance in one chromatogram a wavelength gradient was applied.
2.6.4. Buffer pH
Various trials on the pH of the Potassium dihydrogen phosphate buffer were made to achieve the optimum pH at which all API peaks are well separated. For the optimum peak shape and peak tailing, a buffer pH of 3.0 was selected.
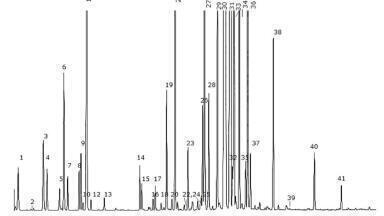
The chromatographic conditions were optimised with a mobile phase of pH 3.0 Potassium dihydrogen phosphate buffer and acetonitrile mixture at gradient wavelength with a flow rate of 1.5 mL/min at 40ºC column temperature and 40 µL injection volume. The typical HPLC chromatogram (Figure 1) shows a satisfactory separation of SX and FP.
2.7. Method Validation
The validation of the developed method was performed in accordance with the ICH Q2 (R1) guideline [8] for the following parameters: specificity, linearity, limit of quantitation (LOQ), accuracy, precision, robustness, and solution and mobile
phase stability.
2.7.1. Specificity
Specificity is the ability of the substance to be analysed to be precisely determined in the presence of the matrix effect and additives ensuring the identity of the analyte(s) of interest.
Blank solution, placebo solution, standard solution and assay test solution were analysed and peaks from each of the solutions were determined for SX and FP. Moreover, the impurities in Table 1 were injected to the system in order to determine their relative retention times in the chromatogram hence preventing interfering peaks. With the given chromatographic conditions the PDA detector was used to detect the peak purity.
For a peak to be considered pure, purity angle must be less than purity threshold. SX and FP peaks in the chromatogram were found to be pure as it is seen in Table 2. All compounds were completely separated and no drift in analyte retention time was observed.
2.7.2. Linearity and range
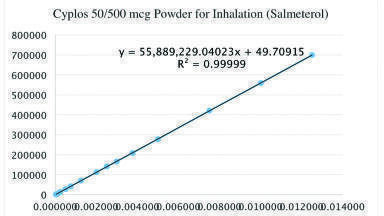
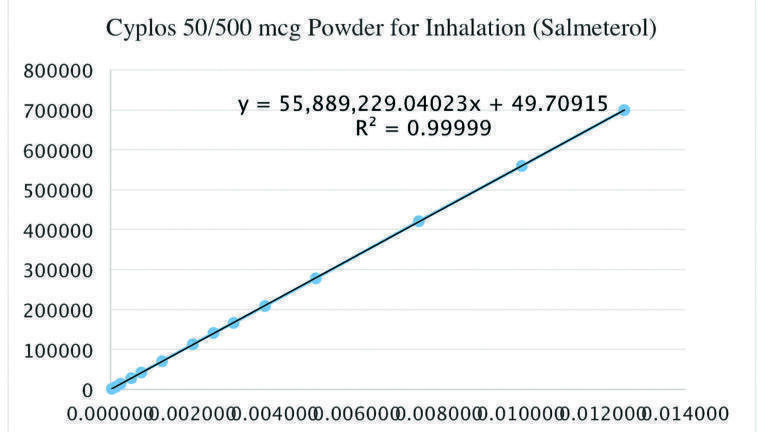
Fourteen different concentrations of standard solutions were prepared to test the linearity range. The calibration curve was plotted as peak area versus concentration of the standard solutions.
The nominal concentration of test solutions for SX and FP are 0.0025 mg/mL and 0.025 mg/mL, respectively. Relative response factors were determined by preparing standard solutions at different concentration levels ranging from LOQ concentration to 0.0125 mg/mL for SX and to 0.125 mg/mL for FP.
The correlation coefficient (r) should not be lower than 0.998 to establish the criterion of linearity according to Kazakevich and LoBrutto [9]. As can be seen in the Graph 1, the correlation coefficients (r) were greater than 0.998. Therefore, it can be concluded that the analytical method was linear for this concentration range.
2.7.3. Limit of Quantification (LOQ)
LOQ values for SX and FP were determined based on signal-to-noise approach according to ICH guidelines. The results were tabulated in Table 3.
2.7.4. Accuracy
The accuracy of the method was determined by recovery experiments. Recovery studies were carried out with three injections at three different concentrations. Three concentration levels of 80%, 100% and 120% of the specification level of SX and FP were prepared. Three samples were prepared for each level. The experimental results (shown in Table 4) reveal that recoveries were obtained between 80% - 120% for SX and FP.
2.7.5. Precision
For the precision parameter of the method validation, system precision, repeatability and intermediate precision studies were performed. System precision studies were carried out by consecutively injecting the standard solutions for six times. Repeatability was studied by consecutively injection of six test solutions, which were prepared separately. Intermediate precision was carried out by injecting six injections of standard and sample solutions within-laboratory variations: different days, different analysts, and different equipment. The relative standard deviation and difference between two analysts were calculated. The lower RSD % values (<10.00) indicate good precision of the developed method shown in Table 5.
2.7.6. Robustness
To demonstrate the robustness of the method, system suitability parameters were verified by making changes in chromatographic conditions such as change in column temperature ± 5ºC, change in mobile phase ratio ± 10 (v/v), change in flow rate ± 0.1 mL/min., change in buffer pH ± 0.1. The retention time and the difference between the results at normal conditions and modified conditions were calculated. According to these modifications excluding the change in mobile phase ratio, it can be concluded that method was robust over an acceptable working range of its HPLC operational conditions.
2.7.7. Stability
The stability of mobile phase, standard and sample solutions were carried out by keeping the solutions for 28 days for mobile phase and 2 days for standard and sample solutions, and observing for changes in the area and the retention of the peaks. In addition, the standard and test solutions kept refrigerated and at room temperature (RT) were also monitored at the determined periods and the % change calculated and compared a chromatogram of freshly prepared solutions. Relative difference (%) of SX and FP was calculated based on the values of initial conditions and it should be smaller than 2.0%. The results show that the standard solution was stable for 30 hours at 25 °C, stable for 7 days at RT and 14 days when refrigerated. The test solution was found to be stable for 30 hours at 25°C for the assay, and mobile phase was stable for 28 days in ambient conditions.
3. Discussion & Conclusion
Replacing Sodium Dodecyl Sulphate with Potassium Dihydrogen Phosphate makes this method safer in terms of working conditions. In addition, employing UV detection provides simplicity when considering sample preparation compared to FLR detection which is more sensitive to any kind of contamination. Moreover, using the wavelength gradient, produced a maximum response at the wavelength maximum for SX and FP.
To conclude, the validation studies done in accordance with ICH guidelines prove that the suggested method is accurate, precise, robust, specific and selective.
References
1. Anwar, M., El-Haggar, R, Zaghary, W (2015), Profiles of Drug Substaces, Excipients and Related Methodology Chapter Five- Salmeterol Xinofoate, DOI: https://doi.org/10.1016/bs.podrm.2015.02.002
2. Kercsmar, C. (2012) Wheezing in Older Children:Asthma, Kendig& Chernick’s Disorders of the Respiratory Tract in Children 8th Edition, DOI: https://doi.org/10.1016/C2011-0-05011-1
3. McKeage, K., Keam SJ.(2009), Salmeterol/Fluticasone Propionate: A Review of Asthma, Drugs Volume 69, Issue 13, DOI: https://doi.org/10.2165/11202210-000000000-00000
4. USP 41 NF 36 Fluticasone Propionate and Salmeterol Inhalation Powder
5. British Pharmacopoeia (2016), Draft Monograph of fluticasone and salmeterol inhalation powder pre-dispensed vol. 3.
6. Sodium Dodecyl Sulfate SDS , http://www.merckmillipore.com/TR/tr/product/msds/MDA_CHEM-817034?Origin=SERP
7. Potassium Dihydrogen Phosphate PDP; http://www.merckmillipore.com/TR/tr/product/msds/MDA_CHEM-104873?Origin=PDP
8. EMEA, (2005), International Conference on Harmonization (ICH) Q2 (R1): Validation of Analytical Procedures—Test and Methodology
9. Kazakevich Y.V, LoBrutto R. (2007), HPLC for Pharmaceutical Scientists, Wiley