LC-MS
Published over 8 years ago. See the latest and most current information on LC-MS.
Science and Advice for Scottish Agriculture (SASA) is an Official UK laboratory that on behalf of the Scottish Government, participates in statutory UK and EU annual surveillance programmes that monitor UK and imported food & drink for residues of pesticides and their metabolites.
LC/MSMS has been a front-line tool in the laboratory for many years but with older instrumentation it was not possible to generate reliable data for all pesticides in a single run.
A highly sensitive LC/MSMS instrument has been commissioned. The instrument is capable of simultaneous acquisition of hundreds of MRMs incorporating fast polarity switching without compromising data or generation of sufficient data points across each peak for quantitation. However, due to the excellent robustness of the system, it has been possible to modify the generic QuEChERS procedure and omit the clean-up step. Consequently, we directly carry out quantitative analysis of crude extracts.
The above features are demonstrated with examples of quantitative surveillance data, UK and EU Proficiency Test data and system performance data.
Introduction
EU legislation requires member states to carry out post-approval surveillance monitoring of pesticides in food and feed to ensure that residues do not exceed Maximum Residue Levels (MRLs) [1]. A MRL is the highest level of a pesticide that is legally tolerated in or on food and feed. MRLs are not safety limits but the highest level expected when pesticides are used in accordance with good agricultural practice. The regulation covers pesticides currently and previously used in agriculture in or out with the EU. For pesticides not listed, a default MRL of 0.01 mg/kg is applied. The co-ordinated EU multi-annual control programme sets out for each member state the minimum number of samples to test and which specific pesticide and crop combinations to test [2]. Member states appoint official laboratories to carry out this statutory work.
Gas Chromatography coupled with Tandem Mass Spectrometry (GC/MSMS) and Liquid Chromatography coupled with Tandem Mass Spectrometry (LC/MSMS) have been front-line tools in official pesticide laboratories for many years. Tandem mass spectrometry provides the required specificity to detect and quantify hundreds of pesticides in complex matrices. As well as the specificity, the instrumentation must also have adequate sensitivity to comply with the regulations.
Official control laboratories have implemented various multi-residue sample preparation approaches to include many pesticides with different physicochemical properties such as the Quick, Easy, Cheap, Efficient, Rugged and Safe (QuEChERS) extraction technique [3] followed by GC and/or LC coupled with MS. Methods must comply with Method Validation and Quality Control Procedures for Pesticide Residues Analysis in Food and Feed SANTE guidelines [4]. The aim for laboratories is to generate SANTE compliant data for all pesticides in as few analytical runs as possible. With older instrumentation it was not possible to generate reliable data for all pesticides in a single run. Samples often had to be analysed using several different approaches in order to achieve the required detection limits of 10 parts per billion (ppb), to cover all pesticides and to provide sufficient identification points to confirm positive residues. Testing Laboratories must be accredited to international standards (ISO 17025:2005) and as part of this must demonstrate proficiency by participating in several analytical proficiency tests each year.
In this work, a highly sensitive Sciex 6500 (QTRAP) LC/MSMS instrument was used to improve the workflow. The instrument is capable of simultaneous acquisition of hundreds of Multiple Reaction Monitoring (MRM) experiments incorporating fast polarity switching without compromising data and ensuring generation of sufficient data points across each peak for quantitation. The instrument has excellent robustness allowing modification of the QuEChERS procedure, in order to directly analyse crude extracts without the need for a clean-up step.
The above features will be demonstrated with examples of quantitative surveillance data, UK and EU Proficiency Test data and system performance data.
Experimental
Sample preparation
Fruit and vegetable samples submitted as part of the 2016 UK monitoring programme were cryogenically milled using solid carbon dioxide and a R23 vertical cutter mixer from RobotCoupe (UK), Middlesex, UK and stored at -20°C until required for analysis. 10 g of sample were extracted using citrate QuEChERS extraction method [5]. Q-sep QuEChERS salts were purchased from Restek (Thames-Restek), High Wycombe, UK. The dispersive SPE clean-up step was omitted. Sample extracts in acetonitrile with equivalent to 1 gml-1 matrix were filtered through 0.45 µm PTFE syringe filters into vials for subsequent analysis by LC/MSMS.
Pesticides to be detected by LC/MSMS were purchased as neat reference standards of purity ≥ 98% from either QMX Laboratories (Thaxted, UK), LGC standards (Teddington, UK) or Greyhound Chromatography (Birkenhead, UK). Single stock solutions of each pesticide were prepared in-house in methanol at concentrations of approximately 400 µg/ml. Newly prepared single stock solutions were compared against old stock solutions as per SANTE guidelines. Single stock solutions were combined into mixed standards at approximately 5 µg/ml for most pesticides and stored at 4-7°C until required for analysis. Pesticides were grouped into two sets of mixtures (A and B) to safeguard that pesticides known to convert to one another were in separate mixtures, for example thiophanate-methyl and carbendazim, ensuring that quantitative results could be achieved for positive samples without the need for analysis with separate standards. HPLC grade methanol and acetonitrile were supplied by Rathburn Chemicals (Walkerburn, UK). Ammonium acetate was supplied by Fisher Scientific UK, Loughborough, UK. Matrix-matched calibration standards at 5 levels were prepared in appropriate (organic) fruit or vegetable matrix that had been extracted using the citrate QuEChERS extraction method. A mixture of internal standards containing carbendazim D4, methomyl D3 and pendimethalin D5 was added to each sample and standard as an injection internal standard.
A Shimadzu Nexera UHPLC system (Shimadzu, Milton Keynes, UK) was coupled with a Sciex 6500 QTRAP mass spectrometer (Sciex, Warrington, UK). A Genius 3031 nitrogen generator from Peak Scientific, Inchinnan, UK was used to supply gas.
UHPLC Set-up
Run time: 2 x 17 min
Column: Kinetex 2.6 µm, C18,
50 x 4.6 mm with
Security Guard cartridge
(Phenomenex, Macclesfield, UK)
Injection volume: 3 µl
Column temperature: 40°C
Flow rate: 0.4mL/min
Eluent A: Methanol/Water 5/95 v/v + 5mM ammonium acetate
Eluent B: Methanol + 5mM ammonium acetate
Gradient elution:
MS Set-up
Ionisation mode: Electrospray in positive and negative ion mode with polarity switching. Multiple Reaction Monitoring (MRM).
Ion Source Temperature: 425°C
Collision (CAD) gas: High
Ionspray voltage: switching between +4500 and -4500V
Curtain gas: 35 psi
Ionspray gas 1: 60 psi
Ionspray gas 2: 50 psi
Entrance potential: 10 V (positive ion) and -10 V (negative ion)
Results and Discussion
There are many considerations when deciding upon a workflow for pesticide residue analysis. Hundreds of pesticides and their metabolites must be screened for, in complex matrices down to low levels and positive residues must be quantified and confirmed with sufficient identification points in accordance with SANTE guidelines. Detection limits are typically 10 ppb for most pesticides although some pesticides need to have lower detection limits since they have much lower MRLs due to their toxicity. For example the MRL for carbofuran in some crops is 0.001 mg/kg necessitating a detection limit of at least 1 ppb. The desired throughput of the laboratory is also important when selecting the analytical approach.
Most surveillance monitoring samples require to be reported either monthly or quarterly therefore high throughput was not crucial. The aims of the method described here were to obtain fully quantitative, robust results for all pesticides in positive and negative ionisation modes and to generate sufficient confirmation data by acquiring at least two MRMs per pesticide in the first analytical run. In this way samples only needed repeat analysis if residue levels exceeded the calibration range (5 - 80 ppb). Calibration curves in this range (and higher for many pesticides) were linear. Typically for positive samples where residues exceeded 200 ppb these were diluted into the linear range of the instrument.
The position of the probe in the Turbo VTM source can greatly affect the sensitivity of the analysis. It is desirable to ensure that for typical LC flow rates the position of the electrode is not too close to the orifice of the mass spectrometer to avoid undesirable components fouling the system. In this work the probe vertical micrometer position was set to 3 mm and the probe horizontal micrometer position was set to 6 mm.
The use of the advanced Scheduled MRMTM Pro algorithm in Analyst software aided the acquisition of the large number of MRMs whilst still ensuring that there were sufficient data points across each peak (>10 points required for quantitation) even in congested areas of the acquisition method.
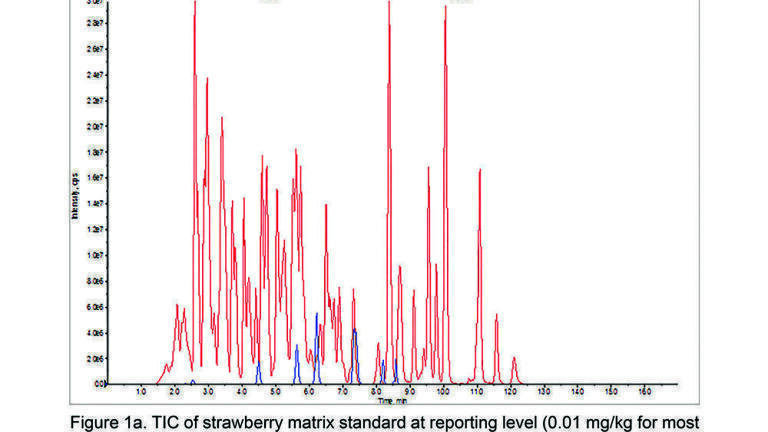
Figure 1a shows the total ion current (TIC) in positive ion mode (red) and negative ion mode (blue) of a strawberry matrix standard at 10 ppb. Figure 1b shows the TIC of a 2016 strawberry sample that contained 16 pesticide residues in the range 10 – 300 ppb. 15 of these pesticides were acquired in positive ion mode (red) and one was acquired in negative ion mode (blue). The extracted ion chromatograms (XIC) presented Figure 1c show boscalid (+MRM) quantifier and qualifier transitions for the strawberry matrix standard at 10 ppb and 2016 strawberry sample containing 16 pesticide residues including boscalid (+MRM) at 80 ppb. XIC of fludioxonil (-MRM) quantifier and qualifier transitions for strawberry matrix standard at 10ppb and 2016 strawberry sample containing 16 pesticide residues including fludioxonil (-MRM) at 100 ppb are displayed in Figure 1d.
The figure is typical of the good peak integrity with excellent signal to noise and sufficient data points in both ionisation modes for all pesticides which was maintained throughout 2016 without the need for remedial maintenance activities. Simple cleaning of the mass spectrometer’s curtain plate was sufficient to maintain this consistent performance without the need to vent the MS for cleaning in between each annual preventative maintenance visit by the service engineer.
Consistent retention times were achieved but Scheduled MRMTM Pro has a function where acquisition of an MRM is extended by up to one minute if the response is still above a threshold at the end of the acquisition window. The Security Guard cartridge was replaced if backpressures increased or peak shapes deteriorated but this was only required after several thousand injections. Column life was also up to several thousand injections of crude extracts from a variety of matrices over approximately 9 months of continuous use.
The methodology was used throughout 2016 to analyse hundreds of fruit and vegetable samples as part of UK and EU monitoring programmes. A summary of the residues quantified and confirmed in 2016 is presented in Table 2.
Figure 2 shows the laboratory’s consistent performance in FAPAS proficiency tests (4 rounds) and EU proficiency tests (2 rounds) for LC/MSMS compounds in 2016. Z-scores must be in the range -2 to 2 to be acceptable. All 37 compounds in these proficiency tests that were analysed using this method had acceptable z-scores.
The consistent performance and stability of the instrument over time i.e. 100 injections using full acquisition method A incorporating polarity switching is illustrated in data presented in Figure 3. Consistent ion ratios were achieved regardless of residue level for confirmation of positive findings.
The method and approach should be future proof in order to cope with most analytical challenges but modifications could be made to solve particular problems and to ensure that data remains SANTE compliant even when analysing very difficult matrices.
The instrument’s fast electronics means that more MRM transitions could easily be incorporated into the acquisition methods without sacrificing the integrity of the data. An additional 9 pesticides were added to method B for 2017 work using the same LC conditions without any compromise in performance. The high sensitivity of the instrument would allow significant dilution of the QuEChERS extracts to ensure that ‘dirty’ matrices would not foul the mass spectrometer.
The linear ion trap was not used in any of the work presented here but this additional functionality already included within the system could be utilised to enhance the method in the future. The QTRAP allows the use of Data Independent Acquisition (DIA) to collect additional MS/MS enhanced product ion (EPI) scans for additional identification. EPI spectra are highly selective and very sensitive due to accumulation of ions in the trap.
Highly selective and sensitive quantitation using MS/MS/MS is another option using the QTRAP, not as part of a routine workflow, but as a tool to solve a particular problem such as matrix interference affecting ion ratios. This is only an option if suitable fragments with enough sensitivity can be obtained.
Conclusions
Implementation of this method has had an extremely positive impact on workflow. Quantitative LC/MSMS data are acquired for hundreds of pesticides in 2 x 17 minute runs with repeat runs required only if positive findings exceed the calibration range. Less than 10 minutes per week are spent on instrument cleaning. Significant time has been saved on data processing due to the excellent signal to noise. The method/system has the capacity for modifications to address future challenges that may be encountered in pesticide residue analysis.
References
1. Regulation (EC) No 396/2005 of the European Parliament and of the Council of 23 February 2005 on maximum residue levels of pesticides in or on food and feed of plant and animal origin.
2. Commission Implementing Regulation (EU) 2017/660 of 6 April 2017 concerning a coordinated multiannual control programme of the Union for 2018, 2019 and 2020 to ensure compliance with maximum residue levels of pesticides and to assess the consumer exposure to pesticide residues in and on food of plant and animal origin
3. Anastassiades M, Lehotay SJ, Stajnbaher D and Schenck FJ, JAOAC Int 86(2) (2003) 412-31
4. European Commission. SANTE/11813/2017 21 –22 November 2017 rev.0.
Guidance document on analytical quality control and method validation procedures for pesticide residues and analysis in food and feed.
5. BS EN 15662:2008. Foods of plant origin. Determination of pesticide residues using GC-MS and/or LC-MS/MS following acetonitrile extraction/partitioning and clean-up by dispersive SPE. QuEChERS-method