Data handling
Published over 4 years ago. See the latest and most current information on Data handling.
Design Space Modeling yields faster production and time to market. The comparison of such models allows for science-driven decision making and risk assessment. The collection of the retrieved data makes it possible to write a Knowledge Management Report which also includes a method robustness evaluation. The results are shortened submission processes and a more transparent communication with regulatory agencies.
Introduction
The pharmaceutical industry is a highly competitive entity with many market participants and often only 1 out of 10 drug developments make it to the market. At the same time, investing into expensive, state-of-the-art, often complex new drug formulations and orphan drugs that commonly are designed specifically for small groups of patients, do not necessarily cover the initial development costs. Likewise, there is a significant chance that the developed pharmaceutical product won’t even reach the final production state, generating a loss to the company [1].
Therefore, the continuous effective support of pharmaceutical development (R&D) and manufacturing quality control (QC) to provide the essential help overcoming hurdles such as increased regulatory burden and increased costs associated with finding and developing new drugs is of utmost importance. Although here it may seem less apparent, but those are the analytical tools with high selectivity and specificity that provide the essential information on the critical quality attributes (CQAs) of the drug product - that affect the drug product’s efficacy, quality and safety for its intended use as addressed in ICH Q6A [2]. In turn, meeting these quality aspects will always be the main drivers for successful pharmaceutical market entry and safeguarding a smooth and reliable pharma production.
Among the other analytical techniques, high-pressure liquid chromatography (HPLC) and in particular, reversed-phase liquid chromatography (RP-LC) is one of the most widely used techniques for the analysis of chemical and pharmaceutical mixtures. HPLC is a relatively young analytical technique, it was early 1967 when the first HPLC instrument was built in Csaba Horváth’s Mason laboratory at Yale University [3]. Since then, there has been a huge development in terms of HPLC instrumentation and columns. The industry has seen the introduction of ultrahigh-pressure, inert, low-dispersion LC-systems with extremely accurate solvent deliveries, near-universal photodiode-array detections (DAD) and simple benchtop mass-spectrometers - which are becoming new industry standards. Column technology has seen the development of highly efficient, sub-2µm particles in various chemistries. These developments have revolutionised Life Sciences and continue to pave the way to apply more powerful gradient elution techniques that contribute in a major way to today’s progress in pharmaceutical, biopharmaceutical and metabolite analysis [4, 5].
Within HPLC, there are great advantages of using gradient over isocratic elution, with the former being applicable to a broad polarity range of analytes and also providing the chromatographer with sharp peaks with improved sensitivity and reduced analysis time. Conversely, compared to isocratic elution, working with gradient elution from the beginning, represents a much higher complexity, where method parameter influences associated with underlying chromatographic interactions may appear to be less trivial. This brings to the necessity of applying methodical approaches, adaption of Quality-by-Design principles and use of advanced modelling tools to manage this complexity.
Regulatory Perspective
Modern (U)HPLC methods need to fulfil not only quality standards in terms of enhanced resolving capability but also high throughput and integrated flexibility for subsequent post-approval adjustments [6]. Analytical methods must be highly adaptable to Lifecycle Management and must always deliver high performance, which is why the whole industry is transitioning towards Analytical Quality by Design (AQbD) approaches.
In this sense, developing methods «with the end in mind» to design quality into the analytical method can offer significant advantages. Having seen the advantages of applying QbD of manufacturing processes, industry practitioners successfully adapted those principles to other areas of life science such as analytical method development. Among others, an aspect of AQbD is to include tolerance limits of the parameters involved along with other systematic elements such as a Design of Experiments (DoE) creating each Design Space. This facilitates risk-, and knowledge-based decision making, which in a long-term can not only minimise but also completely avoid any out-of-specification (OoS) investigations.
As an Analytical Quality-by-Design chapter is not yet available in ICH, the corresponding terminologies and detailed steps remain vague and were interpreted and implemented differently by independent groups. For more guidance, one can however turn on the future terminologies and principles used in the concept paper of the upcoming Q14, which will ask, why a method was developed in a particular way and not otherwise. Most recently, the draft of USP <1220> on Lifecycle Management is also pointing in that direction.
USP <1220> aims to substitute the traditional way of method development and validation by a more structured and holistic approach introducing Method Lifecycle Management. A main drawback of the former was the high number of wet-lab experiments needed while the modelled range remained limited to the studied data points, preventing the study of further parameter dependencies. As suggested instead, analytical method development using a structured Lifecycle Management approach integrates existing chromatographic knowledge as derived from fundamental theories. Such mechanistic modelling by design offers a thorough, holistic understanding and allows for unparalleled modelling versatility. The relevant chemometrical chapter of USP <1039> also highlights the use of mechanistic models [7, 8].
Chromatographic Modelling Tools
Among the many chromatographic theories explaining the reversed-phase retention mechanisms, the Solvophobic Theory of Csaba Horváth has proven to be the gold standard, showing practical uses in other LC modes as well [9]. Applying these principles, computational modelling of designated method parameters (%B-T) began in the 1980ies, when Lloyd Snyder and his co-workers designed algorithms to predict retention dependencies on a programmable calculator. Following this concept, the first chromatography-based modelling software (DryLab I and G) was launched in 1986. It used powerful graphics, so called resolution graphs, to visualise peak movements and critical resolution as per changes of eluent strength (%B) in isocratic elution, gradient steepness and steps in gradient elution, pH, and temperature [10]. Nonlinear factors such as pH, additive concentration, or ternary composition could reliably be calculated on the basis of three input experiments.
In the following case studies, we demonstrate the power of mechanistic modelling approaches in the development of analytical methods in HPLC.
Optimising the Gradient
Modelling gradient elution and model-based flexibility in gradient profile provides many benefits in establishing better peak separation both for small- and large molecule mixtures. The main purpose in gradient elution is ideally establishing a more compact elution profile [11]. Typical approach for achieving this in case of small molecules is the insertion of gradient points (one-step or multi-step gradients), or adding isocratic holds to the gradients to separate peaks, that show more similar chromatographic properties. However, as the molecule size increases, the points of attachments to the stationary phase with the active sites of the columns are in a sudden increase. Large molecules therefore, especially proteins, will show specific ‘on-off’ behaviour, also called ‘bind & elute’ mechanism [12].
As a result, well-managed gradient elution control, and in particular, the so-called ‘elution window stretching’ technique becomes necessary [13, 14].
Another impressive illustration of the power of gradient control is the separation of Cys-linked antibody-drug-conjugates (ADCs), forming definite drug-to-antibody ratios (0-8) in homologous order, where with the software a logarithmic-like profile can easily be set [15].
In-silico Options for Method Transfer
Transferring methods from UHPLC to HPLC in the lab or across company locations, or adopting old ‘legacy methods’ using modern techniques, is often challenging. Although analysts can perform simple, geometrical transfers (scale-up/downs) for an existing method, such an approach misses the systematic context of Design Space Modelling. As a side effect, OoS-cases occur and are reported to be common during the above-mentioned method transfers.
Here, using a holistic approach incorporates the essence of chromatographic dependencies, the separation model is delivering clear answers, on how the design space adjusts if method parameters vary.
Robustness Quantification
Designing methods that deliver consistent analytical results over time are key in smooth pharma production. As defined in ICH Q2 (R1), robustness is the main measure of a method’s capability to resist small but deliberate changes over time. The relevant chapter furthermore suggest assessing robustness as early as during the method development phase - however without mentioning any means of assessment [16].
For methods that were adapted from an external source (i.e. pharmacopeia methods), failures are frequent and likely to show up at later lifecycle stages, in particular during routine use. Hence, in case of an incomplete method understanding, steps taken to avoid or effectively manage OoS’ root cause investigations, can be laborious tasks that will occupy additional resources and may hold up batch releases. Bringing methods quickly back into specification therefore can save tremendous amounts of resources.
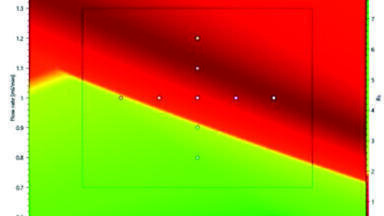
In the modelling context, design spaces can be built for both new and existing methods in order to obtain profound understanding of in-routine behaviour, from which a holistic control strategy (HCS) can be derived. Specifying expected parameter-deviations (tolerance limits), in silico modelling calculates up to 10 HPLC factors at 3 levels each, aligning all relevant components of the separation system. This includes changes of the gradient time, start-, end%B, step height, time, flow-rate, temperature, ternary composition, pH and instrument dwell volume. The software’s modelled results summarise the percentage virtual experiments that pass the robustness threshold. A success rate of 100 % indicates that all modelled experiments pass the Rs,crit.-threshold specified in the analytical target profile (ATP) -typically baseline separation. However, if the success rate is below 100 %, either tighter specification of the parameter-deviations or refinement of the setpoint is needed [17, 18].
Based on the robustness study, meaningful system-suitability-test (SST)-limits can be established with Critical Separation Parameters (CSPs) being simultaneously identified. Next, clear strategies should be set to minimise the effect on variability and to keep separations in specification [19, 18].
Multi-attribute Modelling
Although the critical resolution (Rs,crit) in a chromatographic separation is the most important separation quality attribute, other properties may be needed. For example, in SST specifications, resolution of designated peaks, peak retention times and certain minimum values of peak width or tailing (i.e. to ensure sufficient selectivity between neighbour peaks) may be conducive.
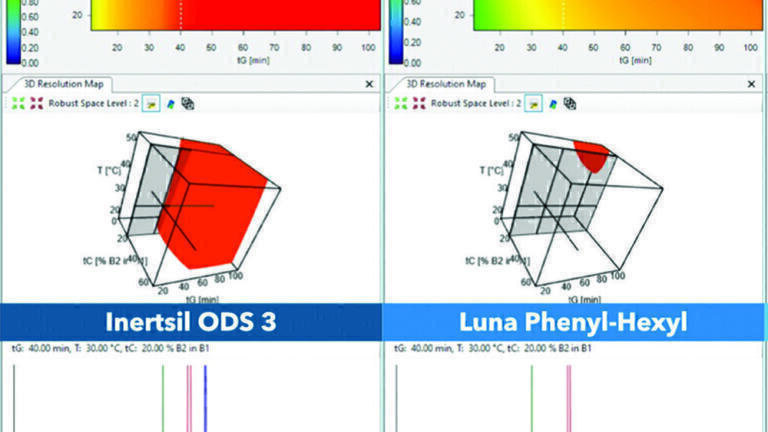
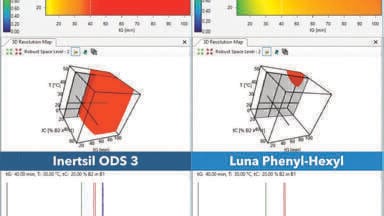
Design Space Comparison
There are three main components in an HPLC-separation system that should be considered for a holistic understanding and judgement of the separation performance: stationary phase, the eluent, and the sample. Indeed, the selectivity provided by the column and the corresponding separation depends greatly on the properties of the studied analyte and the applied chromatographic conditions such as gradient settings, temperature, ternary composition, pH and others.
Similarly, additional influences like column batch-to-batch variations and system-to-system differences need to be considered. Design space models are therefore ideal tools for the characterisation of separation systems, while minimising the experimental work required [17, 20].
Automation
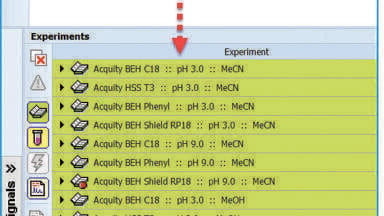
All the examples above can be carried out in an automated fashion using a seamlessly connected software solution. In this fashion, the DryLab suite used above connects directly to Waters Empower CDS. Sample sets for the chosen, Design of Experiments (DoE) are created with only one click after which all twelve experiments are run automatically by Empower, then acquired by DryLab for the Design Space Modelling. Peak tracking benefits of the DryLab-Empower-Connection by means of orthogonal peak tracking: if Mass Spectrometers (QDa or SQD) are available, additional peak information is presented to the analyst in addition to the UV data. For best interpretation of the chromatograms, base peak values are shown in the peak tables. Selected ion chromatograms and mass spectra of selected peaks can be called additionally to the UV chromatograms. After the assessment of the model’s robustness, confirmation runs around the setpoint are seamlessly transferred to Empower where they are executed to then be retrieved by DryLab. By implementing such automated workflows, the reliability can be increased while human, inter-operator errors can be eliminated [21].
Conclusion
The described DryLab Design Space Modelling approach moves away from traditional method development routines based only on statistics. Instead, and in line with the recent ICH Q14 stimuli and USP<1220>, the model yields a profound understanding of the chromatographic interdependencies within the separation system while a Holistic Control Strategy (HCS) can be derived from it. Design Space Modelling effectively expands the chromatographer’s skillset in his quantitative analytical work and can be implemented on a daily basis using the seamless connection of software packages, in this case DryLab and Empower.
By implementing automated workflows, the development of new and badly needed pharmaceuticals can be developed with faster time to market - to the advantage of the suffering patient.
Acknowledgement
The authors would like to thank Gilead Sciences, Waters Corporation, Gedeon Richter and Egis for their continuous collaborative support.
References
1. J. A. DiMasi, H. G. Grabowski and R. W. Hansen, “Innovation in the pharmaceutical industry: New estimates of R&D costs,” J. Health and Economics, 47, (2016) 20-33,.
2. International Council for Harmonisation, “Specifications: Test procedures and acceptance criteria for new drug substances and new drug products: Chemical substances (Q6A),” 6 October 1999.
3. Cs. Horváth, B. A. Preiss, S. R. Lipsky, “Fast liquid chromatography. Investigation of operating parameters and the separation of nucleotides on pellicular ion exchangers”, Anal. Chem. 39 (1967) 1422-1428.
4. F. Tömösi, G. Kecskeméti, E. K. Cseh, E. Szabó, C. Rajda, R. Kormány, Z. Szabó, L.Vécsei and T. Janáky, “A validated UHPLC-MS method for tryptophan metabolites: Application in the diagnosis of multiple sclerosis,” J. Pharm. Biomed. Anal., 185, (2020) 113246.
5. S. Fekete and I. Molnár, Software Assisted Method Development in High Performance Liquid Chromatography, World Scientific Pub Co Inc, 2018.
6. International Concil for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (Q12), “Technical and Regulatory Considerations for Pharmaceutical Lifecycle Management,” 20 November 2019. [Online]. Available: https://database.ich.org/sites/default/files/Q12_Guideline_Step4_2019_1119.pdf.
7. United States Pharmacopeia, “USP <1220> Analytical Procedure Lifecycle (Preliminary draft),” 1 May 2022. [Online]. Available: https://www.uspnf.com/sites/default/files/usp_pdf/EN/USPNF/usp-nf- notices/gc-1220-pre-post-20210924.pdf.
8. N. Matos, M. J. Henson, A. R. Potts and Z. Shi, “Chapter 18 - Guidance for Compendial Use — The USP <1039>Chapter,” in Multivariate Analysis in the Pharmaceutical Industry, Elsevier, 2018, pp. 411-419.
9. C. Horváth, W. Melander and I. Molnár, “Solvophobic Interactions in Liquid Chromatography with Nonpolar Stationary Phases – Solvophobic Interactions Part I,” J. Chromatogr, vol.125, pp. 129-156, 1976
10. J.A. Lewis, J. W. Dolan, L. R. Synder, I. Molnár, “Computer Simulation for the prediction of separation as a function of pH for Reversed Phase HPLC, J. Chromatogr., 592 (1992) 197-208.
11. J. W. Dolan and L. R. Snyder, “Chapter 14 - Method development in liquid chromatography,” in Liquid Chromatography (Second Edition), Elsevier, 2017, pp. 375-388.
12. S. Fekete, J.-L. Veuthey and D. Guillarme, “New trends in reversed-phase liquid chromatographic separations of therapeutic peptides and proteins: Theory and applications,” J. Pharm Biomed. Analysis, vol. 69, pp. 9-27, 2012.
13. E. Tyteca, J.-L. Veuthey, G. Desmet, D. Guillarme and S. Fekete, “Computer assisted liquid chromatographic method development for the separation of therapeutic proteins,” Analyst, vol 141, no. 19, pp. 5488-5501, 2016.
14. S. Fekete, I. Molnár and D. Guillarme, “Separation of antibody drug conjugate species by RPLC: A generic method development approach,” J. Pharm. Biomed. Anal., vol. 137, pp. 60-69, 2017.
15. B. Bobály, G. M. Randazzo, S. Rudaz, D. Guillarme and S. Fekete, “Optimization of non-linear gradient in hydrophobic interaction chromatography for the analytical characterization of antibody-drug conjugates,” J. Chromatogr., vol. 1481, pp. 82-91, 2017.
16. International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use, “ICH Q14: Analytical Procedure Development and Revision of Q2(R1) Analytical,” 2018. [Online Available: https://database.ich.org/sites/default/files/Q2R2-Q14_EWG_Concept_Paper.pdf.
17. D. Enesei, I. Kapui, S. Fekete and R. Kormány, “Updating the European Pharmacopoeia impurity profiling method for terazosin and suggesting alternative columns,” J. Pharm. Biomed. Anal., vol. 187, pp. 1-10, 2020
18. E. Ferencz, È.-K. Kelemen, M. Obreja, E. Sipos, S. Vida, M. Urkon and Z.-I. Szabó, “Computer-assisted UHPLC method development and optimization for the determination of albendazole and its related substances,” J. Pharm.Biom. Anal., vol. 203, pp. 1-11, 2021.
19. A. H. Schmidt, M. Stanic and I. Molnár, “In silico robustness testing of a compendial HPLC purity method by using of a multidimensional design space build by chromatography modeling — Case study pramipexole,” J. Pharm. Biomed. Anal., vol. 91, pp. 97-107, 2014.
20. N. Rácz, R. Kormány, J. Fekete and I. Molnár, “Establishing column batch repeatability according to Quality by Design (QbD) principles using modeling software,” J. Pharm. Biomed. Anal.,
vol. 108, p. 1–10, 2015.
21. A. Zöldhegyi, H.-J. Rieger, I. Molnár and L. Fekhretdinova, “Automated UHPLC separation of 10 pharmaceutical compounds using software-modeling,” J.Pharm Biomed Anal., vol. 156, pp. 379-388, 2018.
22. R. Kormány, I. Molnár and H.-J. Rieger, “Exploring better column selectivity choices in ultra-high performance liquid chromatography using Quality by Design principles,” J. Pharm. Biomed. Anal., vol. 80, pp. 79-88, 2013.























-(1).jpg)