Chiral
Published over 8 years ago. See the latest and most current information on Chiral.
High performance liquid chromatography was used for the achiral separation of four disubstituted Piracetam diastereomeric mixtures. The suitability of normal and reversed phase conditions was also investigated. The influence of the stationary phase nature on the separation of diastereomeric mixtures was tested. It was established that the nature of alkyl substitution and the distance between the chiral centres in Phenylpiracetam derivatives have a major influence on separation conditions. Only on porous graphitic carbon sorbent Hypercarb was successful in separating all the studied diastereomeric mixtures.
It is well known that, for several decades, the unsubstituted 2-(2-oxopyrrolidin-1-yl)-acetamide one)-1 (Piracetam) has been the base for structural analogues substituted with heterocyclic and acetamide moieties. This has permitted the creation of several drugs (Baclofen, Pramiractem, Oziracetam, Phenylpiracetam, etc.) that have been highly effective in the treatment of central nervous system disorders [1-2]. From the chemical point of view mono substitution of Piracetam leads to its conversion into a racemic mixture of two stereoisomers or a single isomer in the case of asymmetric synthesis. Similarly, disubstituted Piracetams are associated with the formation of racemic mixture consisting of four stereoisomers or two diastereoisomeric pairs. Chemical and biological studies in the area of medicinal chemistry are directly linked with the stereochemical separation of racemic molecules and the isolation of most effective isomers [2].
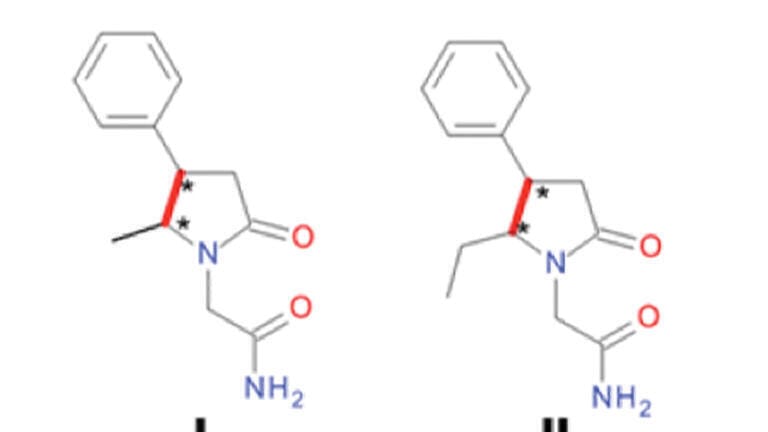
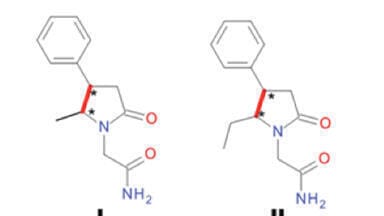
Similar recent studies in our institute have resulted in the synthesis of different alkyl substituted Phenylpiracetams (Figure 1A). The presence of two chiral centres in the molecule of compounds I – IV required the development of a method for the analysis of their stereoisomeric structure. In a previous investigation four stereoisomers of a 4,5-disubstituted piracetam analogue - 2-(5-methyl-4-phenyl-2-oxopyrrolidin-1-yl)-acetamide or compound I (Figure 1B) were separated under chiral LC conditions [3]. Therefore, it is possible for the diastereomeric mixtures of compounds I – IV to effectively monitor the chemical manipulations by qualitative and quantitative analytical control by employing achiral liquid chromatography (LC).
Taking into consideration that a diastereomeric pair consists of two stereoisomers which are not mirror images of one another and therefore have different physical properties, there is the possibility to performing the LC separation under achiral conditions without the use of chiral columns. It is a rather difficult problem because the compounds forming diastereomeric pairs are characterised with minute differences in their physical properties and it is therefore difficult to predict their adsorption behaviour. Even with the popularity of LC as an analytical tool and the large amount of information that has been accumulated about the achiral separation of diastereomers [4-12], understanding the nature of the separation is very challenging. Very few reviews dedicated to the usage of achiral LC for the separation of diastereomeric pairs have been published resulting in separations that are developed based on a trial-and-error approach.
The primary goal of the current investigation was to find suitable achiral LC conditions for the separation of compound I, comprising of two diastereomers E1 and E2 (Figure 1B), providing base line separation (resolution value > 1.5 [13]).
The study consisted of:
a) the suitability of normal (NP) and reversed phase (RP) LC modes;
b) the influence of the stationary phase nature on the separation of diastereomeric mixtures;
c) the use of previously developed separation conditions for other structural types of alkyl substituted Phenylpiracetams II – IV.
2.1. Reagents and chemicals
The corresponding individual diastereomers for compounds I - IV (E1 and E2) were synthesised at the Latvian Institute of Organic Synthesis (Riga, Latvia). HPLC-grade acetonitrile (ACN) was obtained from Merck (Darmstadt, Germany), HPLC-grade methanol (MeOH) and LC grade n-hexane (Hex) was purchased from Sigma-Aldrich (Taufkirchen, Germany). Anhydrous ethanol (EtOH) was obtained from Jaunpagasts Plus (Jaunpagasts, Latvia) and 2-propanol (i-PrOH) from Latvijas Kimija (Riga, Latvia). Phosphoric acid (PA), HPLC-grade methyl tert-butyl ether (MTBE), tetrahydrofuran (THF) and void time markers uracil and 1, 3, 5-tri-tert-butylbenzene was purchased from Acros Organics (Geel, Belgium). HPLC-grade 1,4-dioxane (Diox) and 1-propanol (n-PrOH) was obtained from Fisher Chemicals (Loughborough, UK). Deionised water (R ≥ 18M Ω/cm, TOC ≤ 3ppb) was produced by Milli-Q system (Millipore, Darmstadt, Germany).
Chromatographic analyses were carried out using Waters Alliance (Waters Corporation, Milford, MA, USA) LC systems fitted with a quaternary pump, a degasser, an autosampler, a column thermostat and a Waters 2489 dual λ absorbance. Analyses were controlled by Waters Empower 2 software.
The separations were performed on the columns given in Table 1. All columns were packed with 5 µm particles unless stated otherwise. The column void volume was determined using 1,3,5-tri-tert-butylbenzene (NP) and uracil (RP). Hexane or methyl tert-butyl ether with ethanol, 1-propanol, 2-propanol, tetrahydrofuran or 1,4-dioxane mixtures were used under NP conditions. Different acetonitrile, methanol, ethanol, 2-propanol or tetrahydrofuran mixtures with water or 0.1% phosphoric acid were used as mobile phases under RP conditions. The proportion of each mobile phase component was always measured by volume. Equilibration time is defined as 30 column volumes.
The chromatographic runs were performed at flow rate 1.0 mL/min, and a column temperature of 25°C (NP) and 40°C (RP) if not mentioned otherwise. Detection was accomplished via measurement of the UV absorption at 210 nm. The injection volume was 10 µL. All analytes were dissolved in the mobile phase before injection. The analyte sample concentration was 0.1 mg/mL for E1 diastereomer and 0.05 mg/mL for E2 diastereomer to established elution order.
3.1. Effect of temperature
According to the data in Figure 2 column temperature affects the separation of stereoisomers E1 and E2 (Figure 1B) represented by diastereoisomeric compound I.
The effect of increasing the temperature from 25°C to 60°C (Figure 2) on the separation performance using NP (column Ultrasphere Silica, mobile phase EtOH/Hex 15:85, v/v, %) and RP (column Apollo C18, mobile phase ACN/H2O 20:80, v/v, %). In both modes, the increase of temperature led to the reduction of retention time for stereomers E1 and E2. It was also noted that a decrease in separation occurred due to the increase of temperature (the value of α varied from 1.11 to 1.08) only under NP mode (Figure 2A). In the case of RP (Figure 2B) the increase of temperature resulted in the slight improvement of the separation factor from 1.05 to 1.06. Therefore, in further experiments the following column temperature conditions were chosen: 25°C and 40°C for NP and RP separation modes.
It is well known that changes in the mobile phase (MP) composition can alter the separation on a specific column. The influence of the concentration and nature of mobile phase modifiers on retention, separation factor and resolution of E1 and E2 is given in Table 2. The best separation of diastereomers was achieved using NP conditions. In all experiments E1 diastereomer eluted first under NP mode. A reversed elution order, with E2 diastereomer eluting first, was observed under the RP conditions.
A decrease of ethanol concentration from 35% to 15% (NP mode; column Luna Silica; mobile phase EtOH/Hex; values of kE1 range from 1.62 to 5.62) resulted in the negligible increase of α value. Table 2 shows that a Rs value > 1.5 was achieved under NP mode when the separation factor value > 1.12.
A decrease of the acetonitrile concentration from 35% to 15% (RP mode; column Apollo C18; mobile phase ACN/H2O; values of kE2 range from 1.16 to 12.57) also improved the separation factor and resolution. Unfortunately, under RP conditions the best α value was only 1.07 (Rs = 1.47).
It can be seen from Table 2 that the nature of the MP influences the separation. Replacing EtOH/Hex with EtOH/MTBE as a MP did not give any improvements in separation quality. Moreover, there was observed a decrease in resolution (Rs = 1.25). The replacement of EtOH with i-PrOH for mobile phases containing MTBE resulted in an improved separation (α = 1.17), but negatively affected resolution (Rs = 1.06). The usage of i-PrOH or n-PrOH as n-hexane modifier slightly improved resolution (Rs values 1.53 and 1.73 respectively). There was observed negative effect of THF or 1,4-dioxane on the separation of diastereomers E1 and E2. The best resolution was achieved with mobile phase EtOH/Hex (Rs = 2.20).
Under RP mode both acetonitrile and methanol were suitable for separation of diastereomers with α ~ 1.06 and Rs ~1.2 (Table 2). The use of 0.1% phosphoric acid instead of pure water did not influence the separation and only slightly improved resolution (Rs ~1.3). The effect of THF or EtOH and especially n-PrOH or i-PrOH as MP modifiers was negative.
Separation factors and resolution values for compound I diastereomers (E1 and E2) using different columns (see Table 1) under NP (EtOH/Hex) and RP (ACN/H2O) modes (mobile phases selected so that 5 < kfirst eluted diastereomer < 6) are represented in Figure 3. Five different silica columns (Table 1, columns #1 - #5) were selected for this investigation. It can be seen that the silica type (A, B or C [14,15]) had little effect on the separation factor. The use of a LiChrospher Diol column did not demonstrate any improvement (α = 1.09). Baseline separation was not achieved using cyano and amino columns.
C18 stationary phases are the most widely used reversed phase in LC for different diastereomeric mixtures due to their high effectiveness [4, 9-11]. Figure 3 shows the results of seven C18 sorbents based on different types of silica (Table 1, columns #9 – #15) none of which provided successful results in compound I diastereomers E1 and E2 separation (α = 1.05 – 1.07). Interestingly, in the case of stationary phases with a polar embedded group (PEG) introduced in alkyl ligand (Ascentis RP Amide, Zorbax Bonus RP and Ultra IBD), the selectivity factor decreased (α = 1.04 – 1.05), and a similar poor separation was observed when using aromatic (ACE Phenyl) and fluorinated (Fluophase) sorbents. The Allure PFPP (pentafluorophenylpropyl type sorbent) column gave a separation factor value of 1.08 (Rs = 1.85), and using a 250 mm long column Acclaim C30 (C30 type sorbent) a Rs > 2 was achieved. Among the C18 sorbents Rs > 2 is only observed on Kinetex C18 column (2.6 µm; core-shell type sorbent).
Porous Graphitic Carbon (PGC) sorbents show unique retention behaviour, which differs from that of silica-based stationary phases, and principally are used for the separation of compounds with similar structure, including positional isomers and stereoisomers due to their different interaction with the PGC planar structure [16-18]. Using a Hypercarb column filled with PGC sorbent, the best separation of diastereomers E1 and E2 (α = 1.20; Rs = 3.33) was achieved.
Compounds I – IV differ from each other by the chiral centre position and/or alkyl chain length at the chiral centre (Figure 1A). Diastereomeric compounds I and II were represented by 5-alkyl (Me or Et) substituted in heterocycle 2-(4-pheny-2-oxopyrrolidin-1-yl)-acetamides, but III and IV –by Phenylpiracetams substituted in the acetamide moiety by Et or n-Pr groups.
Under NP mode better separation of compound I diastereomeric mixture was achieved on a Silica C column (α = 1.12) with mobile phase EtOH/Hex. The same chromatographic conditions were chosen for the LC analysis of compounds II – IV. Table 3 shows that increasing the distance between the chiral centres (II vs. III) only slightly improved their resolution. The difference in the size of alkyl substituents in the same chiral centre (I vs. II; III vs. IV) was important only in the case of their introduction into acetamide moiety. Respectively n-Pr substituent improved separation characteristics of compound IV (α = 1.38; Rs = 7.47).
A classical C18 sorbent (Apollo C18), a sorbent with pentafluorophenylpropyl ligands (Allure PFPP) and PGC (Hypercarb) were all used in RP mode with an ACN/H2O mobile phase. The first two columns show similar chromatographic behaviour. The increase of distance between the chiral centres (II vs. III) improved resolution. However, the replacement of Me group by Et in heterocycle (I vs. II) or Et group by n-Pr in acetamide moiety (III vs. IV) resulted in the decrease in separation effectiveness. An increase in resolution caused by the replacement of the Me group with an Et group in pyrrolidin-2-one heterocycle was only observed in the case of Hypercarb column. Moreover, all diastereomeric mixtures under study were only separated with high resolution (Rs > 3) using the PGC sorbent.
A resolution > 2 for the list of diastereomeric mixtures under normal phase mode was achieved using bare silica and EtOH/Hex as the mobile phase. Under reversed phase mode better separations were observed using the Allure PFPP column with ACN/H2O mobile phase. However, using the same stationary phase a baseline separation was not achieved in the case of 5-ethyl substituted Phenylpiracetam (compound II). The PGC column, Hypercarb, separated all of the explored diastereomeric mixtures and it was found that the nature of alkyl substituents and their position in Phenylpiracetam molecule play a major role in the resolution of appropriate diastereoisomers.
1. A.G. Malykh, M.R. Sadaie, Drugs 70 (2010) 287.
2. G. Veinberg, E. Vavers, N. Orlova, J. Kuznecovs, I. Domracheva, M. Vorona, L. Zvejniece, M. Dambrova, Chem. Heterocycl. Compd. 51 (2015) 601.
3. H. Kazoka, O. Koliskina, G. Veinberg, M. Vorona, J. Chromatogr. A 1281 (2013) 160.
4. J. Fekete, M. Milen, L. Hazai, L. Poppe, Cs. Szantay, A. Kettrup, I. Gebefugi, Chromatographia 57 (2003) 147.
5. T. Wang, J. Chromatogr. A 762 (1997) 327.
6. L.Vodeb, B. Petanovska – Ilievska, Acta Cromatographica 17 (2006) 188.
7. E. Mariussen, M. Haukas, H.P.H. Arp, K.U. Goss, A. Borgen, T.M. Sandanger, J. Chromatogr. A 1217 (2010) 1441.
8. T. Toyo’oka, Y.M. Liu, N. Hanioka, H. Jinno, M. Ando, K. Imai, J. Chromatogr. A 675 (1994) 79.
9. R. Bhushan,V. Kumar. J. Chromatogr. A, 1190 (2008) 86.
10. M. Hoshino, E. Matsui, K. Yajima, A. Okahira. J. Chromatogr. A 664 (1994) 104.
11. X. Gao, W. Chen, G. Zhu, R. Yi, Z. Wu, P. Xu, Y. Zhao, J. Chromatogr. A 1218 (2011) 1416.12] D. Sykora, I. Vins, P. Hermann, F. Kesner, J. Chromatogr. A 665 (1994) 59.
13. L.R. Snyder, J.J. Kirkland, J.L. Glajch. Introduction to modern liquid chromatography, John Wiley & Sons, New Jersey, 2010.
14. J.J. Kirkland, C.H. Dilks, J.J. DeStefano, J. Chromatogr. 635 (1993) 19.
15]. J.J. Pesek, M.T. Matyska, J. Sep. Sci. 32 (2009) 3999.
16. Q.H. Wan, P.N. Shaw, M.C. Davies, D.A. Barrett, J. Chromatogr. A 697 (1995) 219.
17. B. Dethier, M. Laloux, E. Hanon, K. Nott, S. Heuskin, J.P. Wathelet, Talanta 101 (2012) 447.
18. N. Apel, E. Uliyanchenko, S. Moyses, S. Rommens, C. Wold, T. Macko, K. Rode, R. Brull, J. Chromatogr. A 1488 (2017) 77.


















-(1).jpg)

2.jpg)