LCxLC
Published over 8 years ago. See the latest and most current information on LCxLC.
This report demonstrates comprehensive 2D-LC in which two mass spectrometers in parallel plus four other detectors (UV, FLD, CAD, and ELSD) are used to monitor the first dimension (1D), while two more mass spectrometers in parallel plus a UV detector are used to monitor the second dimension (2D). LC1MS2 x LC1MS2 = LC2MS4 was employed to identify and quantify tocopherols, diacylglycerols (DAGs), and triacylglycerols (TAGs) in Jacaranda mimosifolia seed oil (JMSO) for the first time. Non-aqueous reversed-phase (NARP) HPLC in the 1D was coupled to ESI-MS and APCI-MS employing selected ion monitoring and selected reaction monitoring (SRM). We found 591.0 ± 13.5 μg/g of α-tocopherol and 517.6 ± 7.8 μg/g of α-tocopherol in JMSO by SRM APCI-MS and report the percent relative compositions of DAGs and TAGs. We used a lab-made silver-ion column for UHPLC in the 2D coupled to APPI-MS and ESI-MS to identify cis/trans isomers and regioisomers.
Introduction
One of the fastest growing areas of chromatography today is two-dimensional liquid chromatography (2D-LC, or LC × LC). This burgeoning technique provides greater overall separation efficiency (as judged by peak capacity), because the peak capacity of a 2D separation is theoretically the product of the individual 1D peak capacities of the two component dimensions [1]. Although this ideal multiplicative peak capacity is rarely achieved in practice, it is clear that 2D-LC separations can provide greater resolving power than 1D-LC alone. The theoretical aspects of 2D-LC have been described extensively in recent years, and these reports should serve to convince the reader of the value of these types of separations. Applicable terms, a specific nomenclature, and conventions have evolved with the development of LC × LC, as described by Marriott et al. [2] and Schoenmakers et al. [3].
Most 2D-LC experiments reported have similar overall characteristics. Most employ one or more detectors (e.g., UV and mass spectrometry, MS) at the outlet of the second dimension (2D) to monitor the elution after both dimensions of separation. The chromatographic profile of the first dimension (1D) is reconstructed from multiple slices, or samplings, across each peak. Too few slices across a peak results in ‘under-sampling’, which does not adequately reproduce the profile of the 1D. Thus, the time allowed for each 2D separation is the 1D peak width divided by the desired number of slices across the peak (preferably at least 4). For example, a 1 min peak width in the 1D would allow four 15 s 2D separations across its peak width. For this reason, 2D separations often use very high flow rates to accomplish very rapid separations, which requires UHPLC equipment to handle the elevated pressures that accompany such high flow rates and fast-scanning mass spectrometers to obtain as many spectra across the narrow peaks as possible. These 2D conditions are coupled with low 1D flow rates (often from conventional HPLC) to provide wider peaks and low effluent solvent volumes to minimise solvent incompatibility, column overloading, and other issues.
We recently reported the first use of two (dual parallel) mass spectrometers plus four other detectors (a UV detector (UVD), fluorescence detector (FLD), corona charged aerosol detector (CAD), and an evaporative light-scattering detector (ELSD)) for direct detection of the 1D, combined with two more (dual parallel) mass spectrometers plus a UVD in the 2D, in what was described as an LC1MS2 × LC1MS2 = LC2MS4 configuration [4]. While not the first time that a detector had been used to monitor the 1D in 2D-LC [5], this was the first time dual parallel mass spectrometry (DPMS) [6], or LC1MS2, was used in the 1D plus four other detectors, as well as being the first time DPMS was used in the 2D (plus a UVD), for an additional LC1MS2. Since the 1D was monitored directly by six detectors, the peak profile did not have to be reconstructed from the slices across the 1D peaks, thereby bypassing the problem of under-sampling. This approach also allowed accurate quantification of tocopherols using conventional calibration curves, instead of relying on quantification of ‘blobs’ in 2D chromatograms, which is more problematic than quantification by conventional 1D-LC [7]. The LC2MS4 experiments also allowed percentage relative quantification of diacylglycerols (DAGs) and triacylglycerols (TAGs) in Parinari curatellifolia (African mobola plum). The use of high-resolution accurate-mass orbitrap MS in the 1D also allowed identification of previously unreported oxo-DAGs and oxo-TAGs.
That work is replicated and extended in this report, in which the LC1MS2 × LC1MS2 arrangement of instruments is employed for analysis of Jacaranda mimosifolia (blue jacaranda) seed oil (JMSO), which contains jacaric acid (Ja), a conjugated polyunsaturated fatty acid, 8Z, 10E, 12Z octadecatrienoic acid. We provide here the first report of quantification of DAGs, TAGs, and tocopherols in JMSO. In the 2D, we employ a lab-made silver-ion UHPLC column, which separates lipids based on their degree of unsaturation, as well as the location and type of unsaturation (cis versus trans isomers, and differentiation of regioisomers). Thus, isobaric TAG isomers that coelute using NARP-HPLC and cannot be distinguished using high-resolution accurate-mass (HRAM)-MS in the 1D are separated and quantified in the 2D.
Experimental
Sample preparation. Jacaranda mimosifolia seeds were ordered from Sheffield’s Seed Co. (Locke, NY, USA). ~500 mg were ground and extracted using the chloroform/methanol (MeOH) extraction method of Folch et al. [8], with the specific details provided in the supporting information of our previous report [4] noting that the KCl wash was 0.9%, not 0.1% KCl. The oily residues averaged ~20% of starting weight.
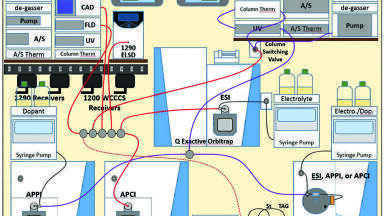
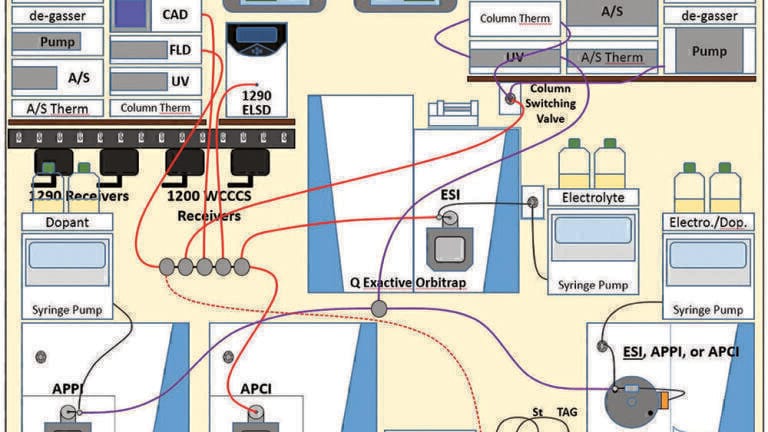
Chromatographic analyses. The arrangement of instruments is shown in Figure 1. An Agilent Technologies (Santa Clara, CA, USA) 1200 HPLC instrument employing two reversed-phase columns in series (Inertsil ODS-2, 25.0 cm x 4.6 mm, 5 µm particles), maintained at 10ºC, was used for the 1D separation. A solvent system of MeOH, ethanol (EtOH), acetonitrile (ACN), and dichloromethane (DCM) was used to accomplish the NARP HPLC separation, using the gradients described previously [4], with 54 min runs for fat-soluble vitamin (FSV) standards and 130 min runs for JMSO samples, at 1 mL/min. A silver-ion UHPLC column was made by loading an ES Industries (West Berlin, NJ) Epic SCX strong cation exchange column (10.0 cm × 2.1 mm, 3 µm) with silver from AgNO3, as already described [4]. The silver-ion column, maintained at 10ºC, was used with a gradient of 1% acetonitrile (ACN) in MeOH to 10% ACN in MeOH, provided by an Agilent Technologies 1290 binary UHPLC at 1 mL/min, as recently described [4]. The fill time of the two alternating 100 µL sample loops on the Agilent G1170A switching valve was 1.86 min (based on the split flow of 53.67 μL/min), with a modulation time of 1.91 min.
In the 1D, the effluent all went through the Agilent Technologies UVD and FLD, since these are non-destructive detectors, and then to a splitter, where the flow was split by a series of tees, where the flow to each detector was dictated by the length and internal diameter of the fused-silica capillaries attached to the outlet of each tee. One branch of a tee went to a corona charged aerosol detector (CAD), one branch went to an evaporative light-scattering detector (ELSD), one branch went to a Thermo Fisher Scientific (San Jose, CA, USA) HRAM Orbitrap mass spectrometer operated in electrospray ionisation (ESI) mode with ammonium formate (via syringe pump) as electrolyte, one branch went to a Thermo Fisher Scientific TSQ Vantage EMR mass spectrometer in atmospheric pressure chemical ionisation (APCI) mode, and one branch went to the G1170A switching valve to the 2D.
In the 2D, the effluent all went through the UVD and then to a single tee, where the flow was equally split to a TSQ Quantum Access Max operated in atmospheric pressure photoionisation (APPI) MS mode with acetone supplied as dopant (via syringe pump) and to an LCQ Deca XP ion trap mass spectrometer in ESI-MS mode with ammonium formate (via syringe pump) as electrolyte.
Specific acquisition parameters for all five LC detectors plus the four mass spectrometers have been provided elsewhere [4]. Acquisition on all instruments was coordinated by the wireless communication contact closure system (WCCCS) described in detail previously [9].
Fatty acid abbreviations (carbon number:double bonds). M: myristic acid, 14:0; P: palmitic acid, 16:0; Po: palmitoleic acid, 16:1; Ja: jacaric acid, 8Z,10E,12Z-18:3; L: linoleic acid, 9Z,12Z-18:2; O: oleic acid, 9Z-18:1; S: stearic acid, 18:0; A: arachidic acid, 20:0; G: gadoleic acid, 20:1; B: behenic acid, 22:0; Lg: lignoceric acid, 24:0.
Results and Discussion
Tocopherols. Figure 2 shows partial UV and FLD chromatograms (22-36 min) of a 2.0 µg/mL standard solution obtained from the 1D separation. These demonstrate that vitamin D3 and gamma tocopherol were overlapped using UV detection, whereas the FLD (an older version borrowed for test purposes) was more specific for tocopherols. Based on these data, we have purchased and installed a newer, more sensitive FLD that will be used for quantification of tocopherols in the future. Because of partial coelution of vitamin D3 and γ-tocopherol and the coelution of α-tocopherol and d6-α-tocopherol, quantification of FSV by UVD is not presented. Instead, selected ion monitoring (SIM) APCI-MS and selected reaction monitoring (SRM) APCI-MS were used for quantification of FSVs, shown in Table 1, since these MS modes easily differentiated overlapped FSVs by mass and were much more sensitive than the older FLD. Extracted ion chromatograms (EICs) from ESI-MS full scan (m/z 200-2000) and targeted scan ranges (m/z 300-750) were also used for quantification of tocopherols for comparison, although the %RSDs using untargeted ESI-MS of un-derivatised tocopherols were too high for reliable quantification. Of course, SRM is the most specific approach, and is not as susceptible to any coincident background or interfering ions like SIM and EICs are, so it is considered the most reliable approach for quantification.
Figure 3 shows the SIM and SRM APCI-MS chromatograms and calibration curves for α-tocopherol ([M+H]+ = m/z 431.389 calc., with SRM → m/z 165.149) in JMSO. These two tocopherols can also be seen in extracted ion chromatograms in Figures 4C (APCI-MS) and 5C (ESI-MS). The time-segmented SIM chromatogram shown in Figure 4C was used for the quantification in Figure 3A, using d6-α-tocopherol as the internal standard (m/z 437.427 calc.).
Both SIM and SRM APCI-MS produced calibration lines with good linearity for α-tocopherol, having coefficients of determination (r2) > 0.99. The selected SRM transitions were highly specific for target tocopherols (especially when combined with the chromatographic retention times) and unambiguously demonstrated their presence in JMSO. SRM results indicated that JMSO contained 591.0 ± 13.5 μg/g α-tocopherol and 517.6 ± 7.8 μg/g α-tocopherol.
γ-tocopherol gave both [M]+· (m/z 416.365 calculated) and [M+H]+ (m/z 417.373 calc.) ions by APCI-MS, which were both monitored by SIM. It was interesting that γ-tocopherol showed a greater ‘first set effect’, in which the SIM produced higher values for the first set of standards in the sequence, which ran before the first sample. The sample gradient runs, which include the separation of TAGs, are longer and include an ACN/DCM gradient. This effect seems to be related to the previously mentioned accumulation of an ACN polymerisation product (‘blob’) on the tip of the corona needle [10]. After the first set of samples ran in triplicate, the remaining four sets of standards gave good linearity by SIM.
Diacylglycerols (DAGs) and Triacylglycerols (TAGs). Analysis of DAGs and TAGs was straightforward, with Figures 4 and 5 representing the total ion current chromatograms (TICs) and mass spectra from APCI-MS and ESI-MS mass spectrometers, respectively, that were used to monitor the 1D of the 2D-LC separation. APCI-MS full-range scans (Figures 4D1, 4E1), data-dependent acquisition (DDA) MS/MS scans (Figures 4D2, 4E2), [DAG]+ scans (m/z 400-750 with 40 V up-front CID), and MS/MS of [DAG]+ (Figures 4D3, 4E3) were used in addition to the targeted analysis of FSV using time-segmented SIM (Figure 4C) and SRM discussed above. For ESI-MS on the HRAM orbitrap™ instrument, full scans (Figures 5D1, 5E1) were followed by DDA MS/MS (Figure 5D2, 5E2), then DDA MS/MS (Figure 5D3, 5E3) of [DAG]+ scans (m/z 350-750) that used 80 V up-front CID to enhance [DAG]+ formation, and finally negative ion full scans (not shown).
Relative quantification of TAGs and DAGs was based on summation of the protonated molecule ions, [M+H]+, for APCI-MS, or ammonium adducts, [M+NH4]+, by ESI-MS, combined with the diacylglycerol-like fragment ions, [DAG]+, formed by losses of fatty acyl chains, [M+H-RCOOH]+. Since DAGs and TAGs differ by sites of unsaturation, which are 2 Da, the 1 x 13C isotopic peaks were included with the [M+H]+, [M+NH4]+, and [DAG]+ peaks to provide more signal for quantification, without loss of specificity, as previously demonstrated [11]. The TAG composition by APCI-MS is given in Table 2 and the DAG composition is given in Table 3. The corresponding compositions by ESI-MS are given in Tables 4 and 5, respectively. As was the case with our recent report on Parinari curatellifolia TAGs, the JMSO samples contained small amounts of oxo-TAGs and oxo-DAGs, which were conclusively identified by HRAM orbitrap™ ESI-MS, although the amounts here were less than those in parinari seed oil. The exact nature of the oxo-TAGs requires additional analysis, such as using NMR, such as was recently demonstrated for analysis of hydroxyl-group containing TAGs in cocoa beans [12].
Four TAGs (LJaL, JaJaL, OJaL, and JaLS) accounted for > 60% of TAG molecular species. The amount of DAGs in Table 3 determined by APCI-MS constituted only 0.80% of the total integrated area, while DAGs by ESI-MS in Table 5 represented 0.97% of the integrated area. To the best of our knowledge, these tables represent the first report of the DAG and TAG compositions of Jacaranda mimosifolia seed oil.
Fatty Acid Composition. The FA compositions calculated from the DAG and TAG compositions are given in Table 6 for comparison of the FA composition determined by MS to that determined by calibrated GC-FID. There was good agreement between the MS results and the FID (converted from weight % to mole %) results. For APCI-MS results, there was less than 4.1% error for each of the six FA present at >0.4% and by ESI-MS there was less than 3.9% error for the FAs present at >0.4%. These results were in surprisingly good agreement to the partial FA composition determined by 13C NMR [13], listed in Table 6, although all saturated FA were determined as a group using that technique. Table 6 represents the most complete FA composition for JMSO reported to date. Other isomers of 18:1, 18:2, and 18:3 were identified using GC-FID, as listed in the last columns of Table 6. Peaks resulting from intact TAGs containing such isomers can be seen in Figures 6 and 7, discussed below. But since there are no TAG standards available to identify these isomer-containing molecular species, we can only report their presence, not their exact identities.
Second Dimension Separation by Ag-Ion Chromatography. Figures 6 and 7 illustrate two-dimensional contour plots and three-dimensional visualisations of JMSO TAGs by APPI-MS (with acetone dopant) and ESI-MS (with ammonium formate electrolyte), respectively. As can be seen, the separations utilise a good proportion of the separation space for TAGs, although FSVs in standard solutions (not shown) were not substantially separated using this binary UHPLC gradient, which was optimised for TAG separation. In unpublished experiments, we have separated vitamin D3 from gamma-tocopherol on the Ag-ion column using a gradient of MeOH/H2O and pure MeOH. Ideally, a quaternary UHPLC instrument would allow the use of one 2D gradient for FSVs and a different 2D gradient for TAGs, but the 2D-LC software will not yet allow it.
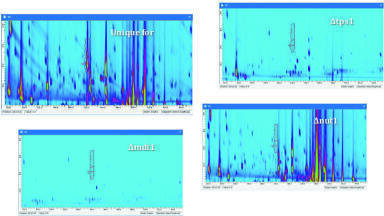
TAG Regioisomers. There are several examples of the separation of regioisomers visible in Figures 6 and 7, such as JaLJa and JaJaL, LLJa and LJaL, JaLP and PJaL, etc. Figure 8 shows EICs for the [JaJa]+ [DAG]+ at m/z 595.5, for m/z 875.7 (the mass of JaJaL) and for m/z 877.7 (the mass of both LLJa and JaJaO). As mentioned in our first report, our method employs a ‘slow’ second dimension separation, in which the modulation time is long compared to the peak width. Instead of having multiple modulations across each peak to minimise under-sampling and then having to reconstruct the 1D from the 2D profiles, we bypass the problem of under-sampling altogether by directly detecting the 1D in detail, as described in the sections above. Therefore, many peaks elute within a single modulation period, and only some peaks are split into two modulation periods when the valve switches in the middle of a 1D peak. This greatly simplifies the appearance of 2D chromatograms and makes quantification of regioisomers much easier. For instance, In Figure 8C JaLJa eluted early in the modulation period, followed by the majority of JaJaL, with the second portion of JaJaL eluted in the following modulation period. The average APPI-MS spectra across these peaks are seen in Figures 8E-8G. The areas of these peaks (Figures 8B+C) were integrated and it was found that the JaLJa peak represented 26.5% ± 1.1% (n=3) of the total area for this TAG (JaLJa + (JaJaL & LJaJa)), while JaJaL (which is not differentiated from its enantiomer LJaJa) represented 73.5% of the molecular species. These proportions are not dramatically different from the statistically-expected ratio of 33%/67%. On the other hand, peaks in chromatograms such as Figure 8D indicated that JaOJa represented only 6.5% ± 0.1% (n=3) of the integrated area, meaning that JaJaO (+OJaJa) represented 93.5% of the regioisomer integrated area.
The physical separation of regioisomers means that they can be quantified without relying on the ratios of fragments in APCI-MS spectra or ESI-MS spectra, with the latter providing slightly less reliable results [14] using that approach than the former. These chromatograms and percentages demonstrate the proof of concept for regioisomer identification using Ag-ion UHPLC as the 2D in a 2D-LC system. Further work should be done using mixtures of pure regioisomers for comparison of the percentages obtained by chromatographic separation to those obtained using a calibration curve approach from the ratios of [DAG]+ ions in mass spectra [15, 16]. The ability of a single chromatogram to elucidate and quantify the regioisomer compositions of TAGs represents a time-saving advantage over running multiple mixtures to construct a calibration line prior to regioisomer quantification.
Conclusions
We have demonstrated again and confirmed our first report showing that comprehensive two-dimensional LC with double dual parallel mass spectrometry (LC1MS2 x LC1MS2 = LC2MS4) can be accomplished using NARP-HPLC in the first dimension combined with an easily lab-made silver-ion column for argentation UHPLC in the second dimension. By using direct detection in the 1D, we have bypassed the problem of under-sampling, and have produced 1D chromatograms that allow both absolute quantification of tocopherols and percent relative quantification of diacylglycerols and triacylglycerols. Using both ESI-MS and APCI-MS for dual parallel MS detection in the 1D, we have been able to simultaneously perform targeted analysis with SIM and SRM for quantification (although SRM is always preferred), plus used high-resolution accurate-mass ESI-MS for identification of oxo-TAGs. These were present in lower amounts in Jacaranda mimosifolia seed oil than they were in Parinari curatellifolia seed oil [4].
By using ‘slow’ comprehensive 2D-LC, we have been able to use older MS instruments that do not have the high scan speeds necessary for faster conventional comprehensive 2D-LC. Furthermore, since peaks are broken into only two modulation periods at most, we have demonstrated the proof of concept that peaks can readily be integrated to determine the percent relative quantification of TAG regioisomers, without the need to analyse multiple mixtures of regioisomers in known compositions. Since more and more labs are realising the extra resolving power possible with two-dimensional LC separations, this work demonstrates additional options for LCxMSy experiments in which mass spectrometry can be used in both dimensions to increase the amount of information provided by the 2D-LC separations.
Acknowledgements
The work of Dr Robert Goldschmidt to extract the oil and perform the analysis of fatty acid methyl esters by GC-FID, and confirmation of identities by GC-MS, is gratefully acknowledged. This work was supported by the USDA Agricultural Research Service. Mention or use of specific products or brands does not represent or imply endorsement by the USDA.
References
1. D.R. Stoll, Introduction to Two-Dimensional Liquid Chromatography – Theory and Practice, in: M. Holčapek, W.C. Byrdwell (Eds.) Handbook of Advanced Chromatography/Mass Spectrometry Techniques, Elsevier/AOCS Press, Champaign, IL, 2017, pp. 250-350.
2. P.J. Marriott, P. Schoenmakers, Z.Y. Wu, LC-GC Eur., 25 (2012).
3. P. Schoenmakers, P. Marriott, J. Beens, LC-GC Eur., 16 (2003) 335-339.
4. W.C. Byrdwell, Anal. Chem., 89 (2017) 10537-10546.
5. D.W. Cook, S.C. Rutan, D.R. Stoll, P.W. Carr, Anal. Chim. Acta, 859 (2015) 87-95.
6. W.C. Byrdwell, Rapid Commun. Mass Spectrom., 12 (1998) 256-272.
7. H.P. Bailey, S.C. Rutan, P.W. Carr,
J. Chromatogr. A, 1218 (2011) 8411-8422.
8. J. Folch, M. Lees, G.H. Sloane-Stanley, J. Biol. Chem., 226 (1957) 497-509.
9. W.C. Byrdwell, J. Lab. Autom., 19 (2014) 461-467.
10. W.C. Byrdwell, Multiple Parallel Mass Spectrometry for Liquid Chromatography, in: M. Holčapek, W.C. Byrdwell (Eds.) Handbook of Advanced Chromatography/Mass Spectrometry Techniques, Elsevier/AOCS Press, Champaign, IL, 2017, pp. 365-405.
11. W.C. Byrdwell, J. Chromatogr. A, 1320 (2013) 48-65.
12. D. Sirbu, M. Corno, M.S. Ullrich, N. Kuhnert, Food Chem., 254 (2018) 232-240.
13. A.P. Tulloch, Lipids, 17 (1982) 544-550.
14. W.C. Byrdwell, J. Am. Oil Chem. Soc., 92 (2015) 1533-1547.
15. L. Fauconnot, J. Hau, J.M. Aeschlimann, L.B. Fay, F. Dionisi, Rapid Commun. Mass Spectrom., 18 (2004) 218-224.
16. A. Jakab, I. Jablonkai, E. Forgács,
Rapid Commun. Mass Spectrom., 17 (2003) 2295-2302.