LC-MS
Published over 10 years ago. See the latest and most current information on LC-MS.
Biotherapeutics are dominating the new pharmaceutical landscape. While liquid chromatography and mass spectrometry (LC-MS) technologies that enable robust characterisation of a protein therapeutic are available, their bioanalysis remains a challenge for laboratories moving a drug product through development. Many techniques can be used for protein quantification; researchers recognise the importance of LC-MS/MS, particularly as it’s become a standard for small-molecule pharmacokinetics.
One of the hurdles of protein quantification is sample preparation. Here we demonstrate a new approach combining pre-measured reagents and proven protocols that can simplify and standardise protein quantification methods. We provide two working examples. First, infliximab is used to demonstrate the achievement of a 10 ng/mL detection limit from 35 µL of plasma, with a step-by-step method that prepares the samples for analysis in less than six hours. Second, we use an antibody-drug conjugate, ado-trastuzumab emtansine, to demonstrate total antibody quantification. These methods ensure both the sensitivity and the transferability required for protein quantification.
In any bioanalytical assay, one of the greatest sources of variability arises from the sample preparation. This can be of particular concern when assays are transferred from sponsor to CRO or from lab to lab, whether within a single company or across multiple sites.
As more drug development efforts focus on large molecule biotherapeutics, such as monoclonal antibodies (mAbs) and antibody drug conjugates (ADCs), traditional ‘small molecule’ bioanalytical scientists find themselves challenged not only by the complexity and time-consuming nature of heterogeneous large molecules, but also by the multitude of potential workflows that exist for protein quantification by LC-MS/MS. Method development time and expertise required are significant. Additionally, in many organisations, the expertise in peptide and protein bioanalysis, if it exists at all, tends to be a rare and valuable skill, held by only a few individuals.
Protein therapeutics such as mAbs and ADCs are on the rise due to their target specificity, lower toxicity, and higher potency. Historically, they have been quantified using ligand binding assays (LBAs), such as the gold-standard ELISA. While these immuno-based methods are sensitive and simple to execute, poor reagent reproducibility, lack of standardisation, cross-reactivity, limited linear dynamic range, and other shortcomings have led the drive to convert to LC-MS/MS. In contrast, mass spectrometry-based methodologies offer many advantages over traditional LBAs, such as multiplexing, broad dynamic range, superior selectivity, and shorter method development times.
LC-MS/MS bioanalytical workflows encompass a multitude of processing segments, each having many steps, and just as complex can be the sample preparation steps that will optimise a large molecule’s quantitative separation and detection. Decisions about specific reagents, as well as the time, temperature, and concentration of the reagents or steps can all impact analytical sensitivity, making it difficult to quickly arrive at a method that produces the desired detection limits.
The difficulty in executing or replicating bioanalytical assays is further aggravated by the complexity of the troubleshooting required when analytical goals such as acceptable accuracy and precision guidelines or reliability and reproducibility standards are not met. Problems that arise in any of those attributes can negatively impact an organisation’s ability to make critical research and discovery-stage decisions.
One of the current trends in quantification of mAbs is to use surrogate peptides. Due to its high sensitivity and specificity, and the increased use of triple quadrupole mass spectrometers in bioanalytical laboratories, the surrogate peptide approach has become the most commonly used strategy for MS-based protein quantification. With this approach, the target protein is digested into smaller peptides. Then one or more unique peptides, surrogate analytes of the target protein, are then analysed by LC-MS/MS.
Proper sample preparation of the target protein and its surrogate peptides is critical to developing a reliable and robust LC-MS/MS assay. By applying a standardised workflow to digest the protein into smaller peptides, laboratories can ensure reproducibility and successful bioanalytical method transfer across labs, analysts, and multiple sites – which is common in the pharma/CRO relationship.
With the infliximab US patent expiration date of 2017 drawing ever closer [1], both CROs and biosimilar research labs are increasing focus on this biotherapeutic. Using a single, universal sample prep method (e.g., Waters® ProteinWorks™ eXpress Digest Kit), an LLOQ of 10 ng/mL infliximab was achieved. (Figure 1).
Infliximab was first immuno-purified from 35 μL rat plasma using a 96-well Protein A agarose-based plate. Samples were then prepared for LC-MS analysis using a commercially available digestion kit and protocol from Waters Corporation. Finally, signature peptides were cleaned-up using a second commercially available kit from Waters based on strong cation exchange of tryptic peptides using SPE [2].
Infliximab samples were affinity purified, digested, and peptides extracted using SPE in less than 6 hours total. This enabled us to begin the LC-MS/MS assay and acquire data the same day, with several 96-well plates being run by the next business morning. (See Appendix for analytical method conditions).
Multiple unique signature peptides as well as a generic human peptides were simultaneously monitored for use in quantification. The best sensitivity was achieved using the unique peptide SINSATHYAESVK from the heavy chain, while additional unique (DILLTQSPAILSVSPGER, light chain) and generic (DSTYSLSSTLTLSK, light chain) infliximab peptides were monitored for confirmation. A unique peptide (SVSELPIMHQDWLNGK) from a common murine mAb standard (Waters Corp, p/n 186006552) was used as the internal standard.
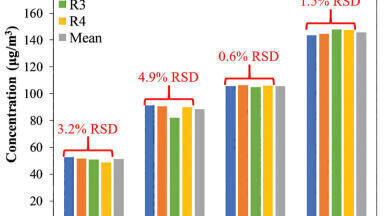
Using the optimised protocol [2] and reagents provided in the kit, only 35 μL of plasma was needed to achieve a detection limit of 10 ng/mL for infliximab (Figure 2). Linearity and accuracy of the standard curves arising from each peptide are summarised in Table 1. The primary, and most sensitive quantitative peptide, SINSATHYAESVK, was linear over 4 orders of magnitude with a mean accuracy (N=3) of >98% for all points on the curve. The additional two peptides were linear over 3.5 orders of magnitude with average accuracies >99% for all curve points.
In addition, the accuracy and precision for the QC samples was excellent with %CVs all <6%. This is summarised in Table 2. In fact, the average %CV for QC samples from the SINSATHYAESVK peptide was ≤3%.
From an assessment of the chromatographic data, it is clear that the quality of the data in terms of peak width and separation from residual endogenous components facilitated both the low level detection and the very high accuracy and precision that were achieved.
Antibody-drug conjugates combine the unique targeting capabilities of mAbs with the cancer-killing ability of cytotoxic drug. Due to their complex and heterogeneous nature, ADCs often require multiple bioanalytical assays to determine efficacy, toxicity, and PK/PD response during drug development stages.
The bottom-up approach, using enzymatic digestion of the ADC/mAb followed by LC-MS/MS analysis, is becoming routine for ADC and mAb quantification. Bioanalytical goals for ADCs include quantification of both the conjugated and unconjugated forms of the ADC, total mAb, cytotoxic payload, and various other catabolites/metabolites. Of the many experiments required to characterise and quantify ADCs, total antibody measurements are important, and are presented in the work herein.
Trastuzumab is a humanised anti-HER2 monoclonal antibody that was approved by the FDA in 1998. With EU patent expiry in July 2014, and impending U.S. patent expiry in 2019, the focus on this drug as well as next-generation potential as an ADC has steadily increased. Ado-trastuzumab emtansine (T-DM1) is an FDA-approved ADC marketed under the brand name Kadcyla, and is used as treatment for patients with advanced breast cancer [3-5]. ADCs, like T-DM1, are composed of cytotoxic small molecule drug (payload) covalently bound to an antibody by a linker.
To prepare standards and quality control (QC) samples, trastuzumab or T-DM1 was spiked into rat plasma at various concentrations (0.1-500 µg/mL). An intact murine monoclonal antibody standard (Waters Corp, p/n 186006552) was used as a generic internal standard.
Using the commercially available kit from Waters and its optional 5-step digestion protocol (which includes reduction and alkylation), a direct digest of plasma (35 µL) containing either T-DM1 or trastuzumab was performed in preparation for LC-MS/MS analysis. Three signature tryptic peptides were used for quantification: IYPTNGYTR, FTISADTSK, and GPSVFPLAPSSK. (See Appendix for analytical method conditions.) While some commercially available kits eliminate reduction and alkylation (we will call this ‘3-step’), the inclusion of reduction and alkylation steps helps ensure detection of conjugated peptides.
From an analytical perspective, tryptic digestion and the choice of a signature peptide pose a challenge for quantification of T-DM1, since it is a lysine-conjugated ADC. Trypsin cleaves peptides on the C-terminal side of lysine amino acid residues, and if a lysine residue is occupied with the cytotoxic drug, cleavage will not occur (‘miscleavage’). Thus, if one were to choose a lysine-containing peptide to quantify T-DM1, there is potential for miscleavage on the lysine residue when it is conjugated with the small molecule drug. Because the signature peptide IYPTNGYTR lacks a lysine residue, one can confidently and accurately use it to quantify both T-DM1 and trastuzumab. For this same reason one would need to be cautious of using the two lysine containing peptides, FTISADTSK and GPSVFPLAPSSK for accurate quantification of T-DM1. Both of these peptides have some degree of small molecule drug occupancy and thus, due to potential miscleavage of the lysine residue, may result in lower calculated concentrations than a non-lysine containing peptide.
For this application, sensitivity, linearity, accuracy and precision data met typical method validation requirements [6]. Standard curves were linear over 3.5 orders of magnitude with the average accuracies of 100% for the standard curve points. For the IYPTNGYTR, FTISADTSK, and GPSVFPLAPSSK tryptic peptides, quantification limits between 0.5-1 µg/mL were achieved. Summary statistics from standard curves for trastuzumab are shown in Table 3.
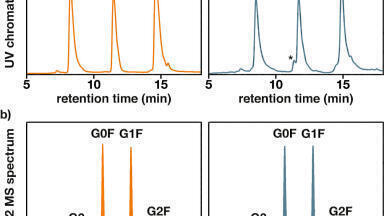
In addition, the accuracy and precision for trastuzumab and T-DM1 QC samples were quantified using the above trastuzumab standard curve specified in Table 3 and were found to be excellent with % CVs <8. The values are summarised in Table 4. Representative QC chromatographic performance and demonstration of sensitive quantification for the IYPTNGYTR peptide is highlighted in Figures 3, Panels A (trastuzumab) and B (T-DM1), respectively.
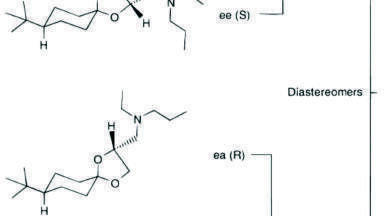
Due to the hydrophobic nature of the cytotoxic drug molecule attached to the antibody and differences in stereo chemical configurations, conjugated T-DM1 peptides generally will elute later in a reversed-phase chromatographic run as diastereomeric pairs. It is also common for T-DM1 peptides to produce a common fragment (547.2 m/z) by collision induced disassociation (CID). This fragment corresponds to part of the drug molecule broken down by the CID process.
In this application, we were successfully able to detect two conjugated, ‘miscleavage’ peptides of T-DM1 (FTISADTSKNTAYLQMNSLR and GPSVFPLAPSSKSTSGGTAALGCLVK) Figure 4, panels A and B illustrate the presence of these conjugated peptides in TDM-1 plasma samples (350 µg/mL), as compared to Trastuzumab (350 µg/mL), and blank rat plasma. Presence of the FTISADTSKNTAYLQMNSLR conjugated peptide was confirmed by multiple MRM transitions, and is shown in Figure 5. Additionally, the area counts from both of these conjugated TDM-1 peptides increased with increasing concentration of T-DM1. This is highlighted in Figure 6, panels A and B.
Commercially available digest and clean-up kits were successfully used to purify infliximab from a typical set of standard curve and QC samples in rat plasma. A limit of quantification of 10 ng/mL was readily achieved, while maintaining excellent linearity and single digit precision. The total sample prep time including an affinity purification step was less than 6 hours. The total digest prep time was just over 2 hours.
Additionally, these optimised kits were successfully used to quantify trastuzumab and the ADC T-DM1 from a typical set of standard curve and QC samples in plasma. Through direct digestion of 35 µL of plasma, quantification limits of 0.5 to 1 µg/mL were achieved, while maintaining excellent linearity, precision and accuracy.
By using these kits, whether the analyst is an experienced or novice bioanalytical and/or biomarker researcher, ultra-low detection limits can be achieved with a simple step-wise protocol and a set of standardised, pre-measured reagents, ensuring both the sensitivity required to make time-sensitive and critical project decisions during drug development.
MRM conditions
Infliximab
Analytical method conditions
LC system: Waters ACQUITY UPLC® System
MS detection: Waters Xevo® TQ-S Mass Spectrometer, ESI+
Data management: Waters MassLynx® Software (v4.1)
Column: Waters ACQUITY UHPLC Peptide BEH C18, 300Å, 1.7 μm, 2.1 x 150 mm
Column temp: 55°C
Sample temp: 10°C
Injection volumes: Infliximab, 10 μL; Trastuzumab and T-DM1, 10 μL
Mobile phase A: 0.1% formic acid in water
Mobile phase B: 0.1% formic acid in acetonitrile
Gradient: MS conditions
Capillary: 3 kV
Cone: 30 V
Source offset: 50 V
Source temp: 150°C
Desolvation temp: 600°C
Cone gas flow: 150 L/hr
Collision gas flow: 0.15 mL/min
Nebuliser gas flow: 7 Bar
1. McKinsey and Company; Data Source: Evaluate Pharma, US Patent Expiration Dates.
2. ProteinWorks Care and Use Guide, Waters Corporation, 2016.
3. Peddi PF, Hurvitz SA. Trastuzumab emtansine: the first targeted chemotherapy for treatment of breast cancer. Future oncology (London, England). 2013;9(3):10.2217/fon.13.7. doi:10.2217/fon.13.7.
4. BarokM, JoensuuH, IsolaJ. Trastuzumab emtansine: mechanisms of action and drug resistance. Breast Cancer Res. 2014 Mar5;16(2):209.doi:10.1186/bcr3621
5. http://www.drugbank.ca/drugs/DB05773
6. FDA Guidance for Industry for Bioanalytical Method Validation, CDER




















-(1).jpg)