LC-MS
Published over 8 years ago. See the latest and most current information on LC-MS.
Requirements regarding the limits of detection of various analytes have become increasingly challenging. In parallel, the sensitivity and resolution of HPLC-MS (high performance liquid chromatography coupled to mass spectrometry) instruments has improved significantly. As a result, all steps of a typical LC-MS workflow must be carried out carefully, utilising the purest reagents available, in order to minimise or avoid sample and system contamination. The quality and handling of solvents, additives as well as consumables (HPLC columns, sample preparation devices) play an important role in maximising sensitivity. This work provides extensive tips and tricks for the proper choice and handling of solvents, additives, HPLC columns, laboratory equipment and systems with a focus on maximising sensitivity.
The prerequisite of any highly sensitive analysis via LC-MS is the use of ultrapure solvents and reagents and careful handling of all associated materials, consumables and systems. This prevents any contamination throughout the entire sample handling process from preparation to MS detection. The overall sample preparation process utilises consumables and reagents such as sample preparation cartridges, pipette tips, solvents and buffers and the preparation and storage of mobile phases. The second step of the process brings HPLC-MS analysis into play; this again includes eluent preparation and storage plus choice of a suitable HPLC column. Crucial steps in these processes are sample and eluent preparation, as both are prone to contamination issues.
In the following sections, various measures and options for maximised LC-MS sensitivity and low limit of detection (LOD) are shown. Each and every tip avoids contaminants causing signal suppression, adduct formation, elevated background noise and increased spectrum complexity.
Typical solvents utilised in LC-MS include water, acetonitrile, methanol, isopropanol or n-propanol. Electrospray ionisation (ESI) is the most widely used ionisation technique and relies on ionising the analyte in solution, evaporating the mobile phase and finally producing single ions. Additives such as acids (e.g., formic acid), bases (e.g. ammonia) or buffers (e.g., ammonium acetate) are used to enable the protonation or deprotonation of the analytes.
The quality of abovementioned solvents (organic eluents or water) as well as additives (buffers, acids, bases) strongly influences the sensitivity of MS detection, therefore utilisation of MS grade solvents and ultrapure additives is mandatory. Make sure that these reagents are labelled as LC-MS grade by the manufacturer.
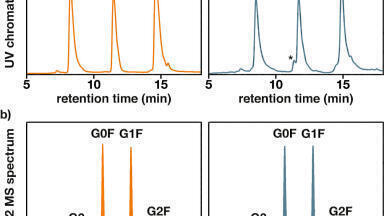
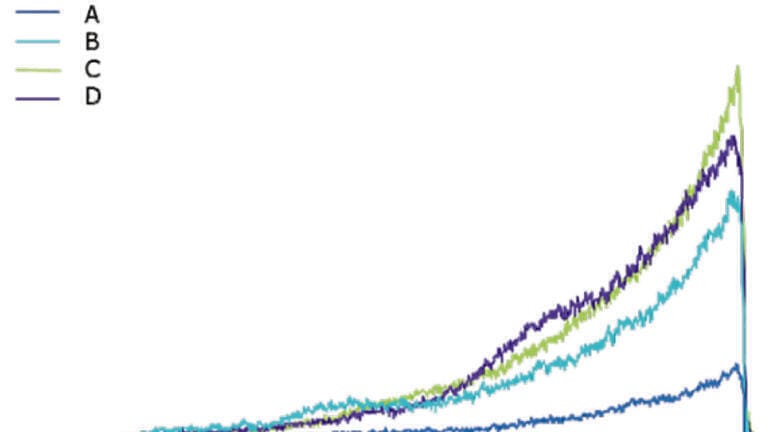
Generally, organic solvents for HPLC, such as acetonitrile and methanol, are available in three qualities: Isocratic grade for liquid chromatography; gradient grade for liquid chromatography, and LC-MS grade for LC-MS gradient runs. LC-MS grade quality solvents should be used for MS analysis, because these show the highest purity and provide lowest signal suppression. Figure 1 displays the results of a reserpine test utilised for the comparison of different acetonitrile qualities and sources. A reserpine solution and a solvent were simultaneously delivered to the MS source, and based on the signal ratio of reserpine to solvent impurities all four solvents were qualified. Another option for testing and comparing of organic solvent qualities is the use of an HPLC gradient run. During such a test, the organic content of the mobile phase is increased from 2 to 100%. When MS intensity is plotted versus time, a ramp is resulting from this setup (Figure 2). The steepness of this ramp can then directly be attributed to the amount of impurities present in a specific organic solvent.
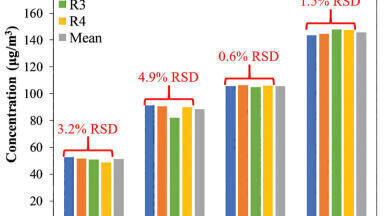
Several sources of HPLC grade water exist, including demineralised tap water, bottled water, and ultrapure water from water purification systems. Demineralised tap water is acceptable for LC-UV runs, but it is not recommended for MS use because of potentially high levels of contaminants. Bottled water and ultrapure water from water purification systems are of comparably high quality. The choice of either of those two options depends mainly on factors such as consumption volume and regularity of use. In case of a comparably low water consumption bottled water is recommended, whereas ultrapure water from a water purification system is suggested in an environment with higher consumption. Water purification systems deliver type I water and are a perfect match with LC-MS analysis. They should be flushed and used regularly in order to maintain or even further improve water quality (Figure 3).
Buffers are utilised to set the pH of a specific chromatographic separation as well as to protonate or deprotonate analytes in solution in support of the electrospray ionisation process. Only volatile buffers and additives such as ammonium formate or acetate or triethylamine should be utilised to adjust eluent pH in LC-MS runs. The use of nonvolatile buffers (e.g., sulphates, phosphates, borates) will cause precipitation in the MS source and ultimately result in tedious cleaning procedures. Ionic strength as the total concentration in the combined eluents, and depending on both buffer concentration and ion charge, should not be set below 20 mM in order to avoid insufficient buffer capacity and irreproducible conditions. If the buffer concentration is above 100 mM, buffer precipitation in organic solvents is possible. This is a general information, which helps to make an HPLC separation work at a first place. However, please keep in mind, that next to adjusting the pH of the mobile phase, the buffer also serves as a support for electrospray ionisation. Consequently, high buffer concentrations might lead to signal suppression. If such a situation is observed, a tradeoff between the quality of an HPLC separation and the extent of signal suppression might need to be made. Alternatively, the stationary phase and/or chromatographic conditions might be adapted, to keep buffer concentration in an acceptable range. The choice of a buffer or acid/base strongly depends on the application. Formic acid works in both positive and negative electrospray mode, and the intensity and sensitivity of the signal is high. For a higher pH, ammonium hydroxide, ammonium acetate adjusted to basic pH or a combination of triethylamine and hydrochloric acid can for example be used. Make sure, that the pH of the eluent matches stationary phase characteristics. For a silica-based stationary phase, the pH of the eluent should be in the range of 2 to 7.5. If the pH is below 2, the bonded stationary phase will detach from the silica backbone and cause an elevated baseline noise; if the pH is set above 7.5, the silica backbone will dissolve causing column voids and bed collapse.
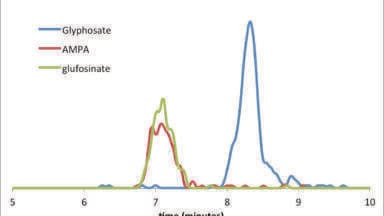
Buffers ionise an analyte molecule M, but the formation of adducts [M+buffer] with, e.g., ammonium, alkali, formate or acetate is possible. This causes additional signals with specific m/z values in a spectrum which may compromise quantitative analyses. Consequently, for samples with high salt load such as food, body fluids or tissue, a desalting step using sample preparation tools like LiChrolut or Supelclean solid phase extraction cartridges is recommended.
Buffers should be prepared by titration of the respective acid and base, as their purity is normally higher as compared to the salts. If any application of salts is necessary, then an MS analysis of those should be performed prior to use to determine, if and what type of contaminant is present in the salts.
Impurities or contaminants of solvents and additives can accumulate on the stationary phase and elute as ghost peaks in gradient runs (Figure 4). This scenario may occur, when the column is equilibrated under highly aqueous conditions prior to a gradient run. Ghost peaks can even appear without equilibration, if the concentration and/or retentivity of contaminants is high and/or if the starting conditions of a gradient are highly aqueous. To avoid ghost peaks in gradient runs, column equilibration time should be kept as short as possible and the flushing volume should not exceed ten column volumes.
Strong acids such as hydrochloric acid, sulphuric acid or nitric acid should be avoided, because they tend to form ion pairs with analytes and therefore make the analyte unsuitable for any type of ionisation. Additionally, some of these strong acids have unfavourable oxidising properties.
Many laboratories tend to use trifluoroacetic acid (TFA) in order to form ion pairs with peptides and proteins and to improve subsequent HPLC separation; however, TFA causes strong ion suppression of the analyte during MS detection and may contaminate the mass spectrometer. If the use of TFA is necessary, then a weak acid or isopropanol should be added to help decrease the signal suppression effect. Alternatively, difluoroacetic acid is an option that decreases the signal suppression effect (as compared to using TFA).
Solvents should be stored in the original manufacturer’s bottle; this can be either surface treated amber or borosilicate glass. Adjustment of the bottle size to specific needs is recommended, because decanting as a source of contamination should be avoided wherever possible. If decanting of a solvent is necessary, borosilicate glass is the commercially available option. Do not utilise any standard clear or soda-lime glass bottles, as alkali and silica can be leached out of the container by the solvent (especially by aggressive, deionised water) and subsequently can form adducts with analytes.
Bottles have to be sealed and connected to the HPLC system using professional caps, adapters and tubing directly mounted to the solvent bottle. Any homemade solution will likely cause contamination of the solvent or eluent and could lead to the evaporation of organic solvents into the lab atmosphere.
Solvents, that are handled or stored utilising plastic devices such as bottles, funnels, beakers or gloves, will leach additives like plasticisers, anti-static agents, stabilisers or anti-slipping agents out of the plastic (Figure 5). Consequently, the use of any plastic device should be avoided during handling and storage of solvents and mobile phases. The only exception are devices that have been tested for leachables and extractables by the manufacturer, e.g., pipette tips or syringes.
Cleaning of laboratory equipment and vessels can most simply be done by evaporation in a fume hood. This is a straightforward method, as all reagents used in MS applications are volatile and of high purity. In cases where contamination is observed, flushing with MS grade solvents will be necessary in order to properly clean the equipment.
Avoid any dishwashing of equipment: Under the harsh, basic conditions, silica and alkali are leached out of glass surfaces and adduct formation or signal suppression is likely. In addition, the dishwashing process leads to the deposition of detergents from the cleaning process and plasticisers from detergent bottle on the equipment surface (Figure 6). If a dishwasher needs to be used for any reason, it is critical, that after cleaning the vessels are flushed with MS grade solvent multiple times.
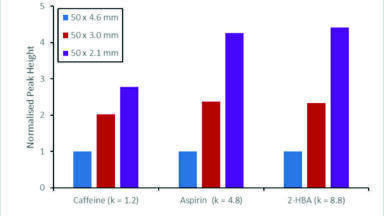
The choice of a specific column dimension is guided by factors such as sample amount, detection technique and necessary loadability, but also by economic considerations such as decreasing solvent consumption. Along with these aspects, attainable sensitivity is also an important issue. Generally, a decrease in column internal diameter (i.d.) – while geometrically scaling injection volume and flow rate accordingly – is a simple means of improving sensitivity of a given separation (Table 1 and Figure 7).
A possible and frequent, but often overseen source of contamination in an LC-MS run is the chromatographic column itself. Most stationary phases are based on a ligand bonded to a particulate or monolithic silica substrate via siloxane bonds. Many of these silica-based bonded phases are inherently prone towards bond cleavage by hydrolysis, mainly at acidic pH (e.g., below a pH of 2). As a consequence, the stationary phase can be stripped off the substrate in the presence of water - a phenomenon referred to as column bleeding (Figure 8).
As a reason of this, non-bleeding columns should be used wherever possible. If no change of stationary phase is possible, e.g. in regulated methods or if a method was established on a specific, but bleeding stationary phase, the use of a washing protocol can help to decrease the negative effect of column bleeding (Figure 9). Alternatively a column should undergo up to ten gradient runs from strongly aqueous to strongly organic before it can be used in MS.
A proper setup of the HPLC system itself can contribute to increased sensitivity as well. An important parameter is the minimisation of dead volume, i.e. the volume of all system parts from the injector to the detector cell, except for the HPLC column volume. Large dead volumes can cause peak broadening, tailing or splitting and lead to poor resolution and decreased performance – and hence can decrease sensitivity and prevent detection of low abundant analytes. Consequently, all system parts (tubing, connectors, fittings) have to provide dead volumes as small as possible.
Replace pump inlet filters every 1 to 2 months or after changing from acetonitrile to methanol (or vice versa) as a solvent. This maintenance will lower the baseline noise and protect the system and column from pump debris.
Eluent filter frits (from solvent inlet filters) should be made out of stainless steel or PEEK rather than glass. Cleaning of the latter is tedious, as buffer residue is hard to remove, and silica and alkali might be leached out of the glass filter and form adducts.
The specific requirements of different chromatographic problems make the use of various mobile phase compositions necessary, ranging from aqueous to organic (Table 2). As a general recommendation, the water content in an eluent used in LC-MS should be set to 5 to 80% in order to work trouble-free and with a stable spray.
If the water content is below 5%, buffers may precipitate in the eluent and the HPLC system. A countermeasure can be the use of a suitable organic solvent or a decrease of the buffer concentration in the eluent. Buffer solubility in utilised solvents should always be checked prior to analysis.
A water content of more than 80% might lead to a breakdown of the MS spray. Several options help to keep the MS spray working.
1. Decrease of the surface tension of the eluent by addition of a volatile organic solvent such as acetonitrile or methanol to the mobile phase after the LC system and in front of the MS source.
2. Reduction of the flow delivered to the MS by means of a split or column exchange.
3. Manipulation of the MS source conditions (increase of dry gas temperature or flow).
In order to avoid any microbial contamination of both system and mobile phase and a phase collapse, water content of the mobile phase should not be set above 95%. If a highly aqueous mobile phase is necessary, 0.05% sodium azide can be added to the eluent. Alternatively, regular flushing of the HPLC system with organic solvent, preferably isopropanol or methanol, prior to standby, is mandatory. Do not use acetonitrile, because acetonitrile can polymerise and block system valves.
Mass spectrometry is a powerful technique for identification and quantification of molecules within complex mixtures. The success of mass spectrometry strongly depends on reducing contamination throughout the entire sample handling process: from sample preparation to equipment cleaning. An important first step in this process is the use of only the highest quality materials for LC-MS, including solvents, buffers, reagents and columns. A combination of ultra-pure solvents and reagents with contamination-free handling results in maximised LC-MS sensitivity and low LOD.